Abstract
There is increasing evidence demonstrating that children with congenital heart disease (CHD) have a greater risk of developing autism spectrum disorder (ASD) in later life. This review aims to summarise the genetics and pathophysiology underlying both conditions. A PubMed search was performed to identify relevant studies exploring the comorbidities of ASD and CHD. The comorbidities of ASD and CHD can be explained by the influence of common and rare variants that contribute to genetic risks. De novo mutations in chromatin remodelling genes, and common genetic loci in the development of brain and heart in utero, can lead to the co-occurrence of ASD and CHD. Furthermore, there are several cases of syndromic ASD with concurrent CHD presentation. Foetuses with CHD may have abnormal haemodynamic changes and alteration of brain circulation in utero, resulting in impaired development of the brain, and increased risk of ASD. Abnormal brain development or brain injury as observed in MRI studies of infants with CHD may also contribute to the risk of ASD. Children with CHD should have regular neurodevelopmental assessment to screen for ASD symptoms for early diagnosis and intervention.
Key Points
1. De novo mutations in chromatin remodelling genes and common genetic loci in the development of brain and heart in utero can lead to co-occurrence of autism spectrum disorders (ASD) and congenital heart disease (CHD). The presence of CHD has also been reported in patients with syndromic ASD such as Fragile X syndrome, Coffin–Siris syndrome, DiGeorge or velocardiofacial syndrome, Phelan–McDermid syndrome, 16p11.2 deletion syndrome, and CHARGE syndrome.2. Foetuses with CHD may have abnormal haemodynamic changes and alteration of brain circulation in utero, which result in impaired brain development, leading to an increased risk of ASD.
3. Children with CHD should receive regular follow-up to assess neurodevelopment and screen for symptoms of ASD.
INTRODUCTION
Children diagnosed with congenital heart disease (CHD) have an increased risk of developing neurodevelopmental disabilities encompassing language, motor, cognitive and social communications in the long-term impairments.1 There is increasing evidence demonstrating that the deficits in social communication among children with CHD may be potentially linked to the diagnosis of autism spectrum disorders (ASD).2 ASD is a heterogenous neurodevelopmental condition often co-occurring with other congenital disorders such as CHD, significantly impacting social communication and behavior.3 A recent meta-analysis has demonstrated a rapid increasing trend in ASD prevalence over recent decades with an average prevalence of 0.6% worldwide.4 The core features of ASD in children include a deficit in social communication and restrictive and repetitive behaviors.5 The severity of CHD has a positive correlation with the risk of neurodevelopmental disorders, including ASD, attributed to presence of de novo mutations in chromatin remodelling genes, Notch signalling, and cilia function.3
Comorbidities of ASD and CHD may arise due to the underlying genetic or haemodynamic changes during in utero or post-natal cardiac surgery.6,7 Additionally, brain development in infants with CHD may be impaired due to intrinsic epigenetic factors.8 The development of both heart and brain occurs simultaneously, sharing common genetic pathways during foetal development.9 Therefore, abnormal heart development may precipitate neurodevelopmental disorders due to these shared pathways.9 Several studies have established that common and rare variants in ASD and CHD may contribute to the concurrent existence of these conditions.10 Mutations in specific genetic loci associated with both ASD and CHD may explain the associations between the two conditions.11 Notably, previous studies have identified a mere five genes that are overlapped between the 65 ASD risk genes and 66 CHD risk genes.12,13
Potential insult to the development of the brain in infants with CHD may occur during intrauterine life.14 Impairment in cerebral blood flow during in utero, post-natal, or post-operative repair also plays a significant role in the comorbidity of ASD and CHD.15 Anomalous development of cardiac structures with intracardiac or extracardiac mixing can lead to a decrease in cerebral oxygen levels or substrate changes, due to alterations in blood oxygen saturation, thus increasing the risk of impaired brain development in foetuses.15 Neuroimaging studies of infants born with CHD often showed abnormalities of the brain structures which may lead to poor neurodevelopmental outcomes.11 Moreover, infants with complex CHD also exhibit neurobehavioral abnormalities, even prior to undergoing cardiac surgery.16 The variability in brain development alterations among infants with CHD can be attributed to haemodynamic instability during cardiac procedures, potentially leading to intraoperative brain injuries arising from ischaemia or hypoxia.7
As the medical management of CHD and cardiothoracic surgical techniques progress significantly, survival of CHD patients has increased, and many patients are now able to survive to adulthood.17 Therefore, the research direction has shifted towards the long-term neurodevelopmental outcomes to improve patient quality of life.18 Understanding the association between ASD and CHD may aid the clinician in early diagnosis and intervention to tackle neurodevelopmental difficulties. Therefore, the aim of this review is to summarise the genetics and pathophysiology underlying both ASD and CHD.
METHODS
A PubMed search was conducted on 31st November 2023 using the search terms “autism spectrum disorder” OR “Asperger’s Syndrome” AND “congenital heart disease” OR “congenital heart malformation”. Additional studies were identified from the bibliographies of relevant articles.
The abstracts and titles of the articles were screened, and the full paper was examined if the abstracts did not provide sufficient information.
In total, nine articles were included in this review. Table 1 shows the epidemiology of the co-occurrence of ASD and CHD.19–27
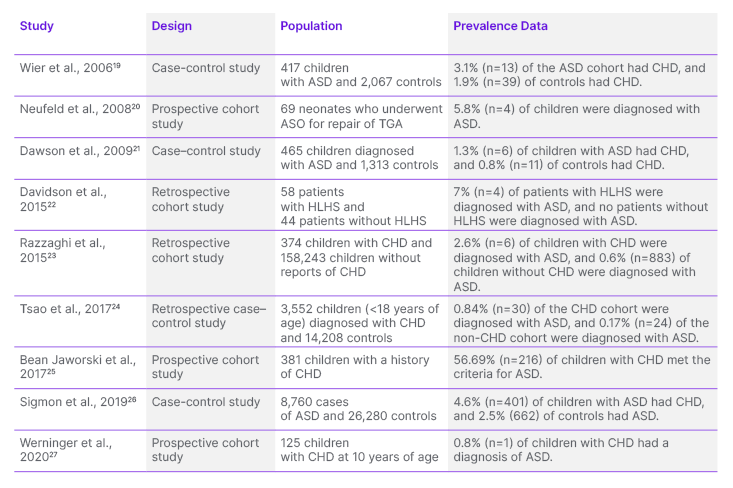
Table 1: Studies exploring the association between autism spectrum disorder and congenital heart disease.
ASD: autism spectrum disorder; ASO: arterial switch operation; CHD: congenital heart disease; HLHS: hypoplastic left heart syndrome; TGA: transposition of great arteries.
GENETICS OF AUTISM SPECTRUM DISORDER AND CONGENITAL HEART DISEASE
Multiple studies have shown that the comorbidity of ASD and CHD may be attributed to common and rare variants, contributing to the genetic risks.10-12,28,29 Genome-wide association studies (GWAS) of FinnGen identified a complex risk locus for developmental left-sided CHD on chromosome 17 near BAHCC1, and associated with LRRC37A2,WNT9B, WNT3, and MYL4 genes.30 Another GWAS study conducted on patients at the German Heart Center, Munich, Germany found a single nucleotide polymorphism located on chromosome 5q22.2 that is shared among all CHD phenotypes.31 Additionally, genes such as MACROD2, GOSR2, WNT3, and MSX1, which are known to play essential roles in embryonic heart development, were implicated in the study.31 On the other hand, GWAS of ASD found that de novo and rare variants of genes such as NEGR1, PTBP2, CADPS, KCNN2, KMT2E, and MACROD2, which are located in the identified loci, were linked to an increased risk of ASD.32
In a study by Rosenthal et al.,10 101 genes with shared genetic risk for both ASD and CHD were identified within a convergent molecular network through network genetics analysis.10 Seven of these identified genes contribute to the genetic pathways of both conditions, including the sodium voltage-gated channel α subunit 2 (SCN2A) which is an ion transport gene.10 These convergent pathways encompass chromatin modifications, mitogen-activated protein kinase/Notch signalling, and ion transport, all integral to the development of brain and heart.10 The rare and common variants genes in these convergent pathways are interconnected within molecular networks.10 In contrast, the study by Satterstorm et al.33 suggests that ASD risk genes lack enrichment in GWAS, implying that common and variant alleles do not significantly influence individual genes.
The association between ion channels and the development of ASD and CHD becomes apparent as different ion-channel genes exhibit genetic signalling via de novo variants within a molecular network.10 In a Xenopus model, a genetic disruption in SCN2A, an ion transport gene, results in major structural defects in both heart and brain development.10 Ion-channel genes may be involved in the regulation of left-right patterning, impacting the left-right patterning in the heart development.34 Other than regulating embryonic development of the heart, SCN2A is also involved in the regulation of cardiac rhythm and post-natal neuron action potential.35 Furthermore, loss-of-function mutations of SCN2A are associated with an increased rate of ASD by 50%.36 SCN2A encodes the voltage-gated sodium channel NaV1.2, expressed in cortical pyramidal neurons and cerebellar granule neurons, which are essential for supporting action potential firing and brain development.37,38 SCN2A mutations result in NaV1.2 haploinsufficiency, due to the production of premature stop codons, contributing to the development of ASD.30 Besides that, de novo mutations of SCN2A variants are also associated with epilepsy and intellectual disability, other than ASD.36
Chromatin remodelling represents a main pathogenic mechanism of ASD.40 ATP-dependent chromatin remodelling involves epigenetic regulation, impacting gene expression and histone-modifying pathways that are essential for embryonic neurogenesis and cardiogenesis.41 Loss-of-function mutations affecting the regulatory functions of histone have implications for both ASD and CHD.42 Mutations in the BAF complex, which alters histone-DNA interactions, have been identified in both ASD and CHD.43,44 The neuronal version of BAF plays a role in the expression of synaptic genes during development of the brain, and its mutation is implicated in ASD.43 Additionally, Smarcd3, which encodes for Baf60c, a subunit of the BAF complexes, is expressed in the heart and is essential for heart morphogenesis.45 Mutation of this gene in a mouse model is associated with impaired development of anterior and secondary heart field and abnormal cardiac muscle development.45 Furthermore, methylation of the H3K4 gene, involved in transcriptional activation, may result in a monogenic form of both ASD and CHD.11,46 Remodelling of H3K4 in the cortex is most active during late gestation to early adolescence, potentially contributing to the development of ASD in CHD patients.47 Another histone-modifying pathway, involving methylation of H3K7 in transcriptional deactivation, is also noteworthy.11 The regulatory protein SMAD2 controls the transcriptional activation process through the removal of a methyl group during embryonic development.39 Furthermore, mutations in CHD8, which are frequently associated with ASD, have also been shown to be mutated in CHD.49,50 Moreover, mutation of CHD7 is also commonly associated with CHD.51 De novo mutations in the H3K4me pathway are involved in the pathogenesis of both ASD and CHD, which lead to the downregulation of transcription of developmental genes in the heart and brain.49,52 A Xenopus model further demonstrated that chromatin remodelling genes such as KANSL1, KAT6A, KMT2A, and KMT2D are involved in the development of both heart and brain.12,33
The study conducted by Assimopoulos et al.53 demonstrated that deletion of 16p11.2 results in more pronounced differences in the structures and functions of the heart and brains when compared to the wild type. This heightened impact is attributed to the presence of multiple genes within 16p11.2 that play significant roles in the development of both heart and brain, such as BCKDK, MAPK3, and SRCAP.53 Additionally, the study also demonstrated the involvement of the Wnt/β-catenin canonical pathway in a convergent signalling pathway crucial for both cardiac and brain development through transcriptional activation.53 Loss of FMR1 protein contributes to increased bone morphogenetic protein signalling, primarily through elevated levels of bone morphogenetic protein type II receptor, affecting Wnt signaling.54 Furthermore, loss-of-function mutations in ARID1B, a target of FMR1 in neural progenitor cells and neurons, result in the upregulation of Wnt/β-catenin target genes.55 β-catenin, in turn, regulates the fibroblast growth factor signalling which is a key element in the regulation of second heart field progenitors during the development of atrial, ventricular, and outflow tract structures.56 In addition, Wnt can also activate a non-canonical pathway involving signalling proteins such as Jun N-terminal kinase, responsible for the regulation the cytoskeleton and cell polarity.57 Both canonical and non-canonical pathways are integral to embryonic cardiac and brain development.11 Furthermore, Assimopoulos et al.53 demonstrated that mutations in the SHANK3 gene cause thickening of the left ventricle anterior wall, which can be explained as they can be effects of CHD, such as tetralogy of Fallot (TOF), pulmonary atresia, or aortic valve abnormalities.
Recurrent microduplications and microdeletions in the distal region of chromosome 1q21.1 are susceptible to variable phenotypes of ASD and CHD.58 Deletion of 1q21.1 is more commonly observed in cases of non-TOF CHD, microcephaly, and schizophrenia, while duplication of 1q21.1 is frequently associated with TOF, macrocephaly, and ASD.59 The duplication of GJA5 in this region results from non-allelic homologous recombination of 1q21.1, leading to an increased risk TOF.59 GJA5 is regulated by Hand2, a cardiac transcription factor, and is expressed in cells of secondary heart field during the development of outflow tract.60 Furthermore, GJA5 also encodes for protein connexin-40, a cardiac gap junction protein responsible for cell adhesion and cell-cell communication.59 Experimental studies in mice with knockout connexin protein showed increased rates of CHD and brain malformations.61 Furthermore, several genes in the 1q21 critical region are expressed in the brain, including HYDIN2 and three NOTCH2NL genes.62 NOTCH2NL genes are essential for cortical development, where overexpression of the genes delays neuronal differentiation, while deletion of the genes accelerates it.63 Loss of genomic stability of these genes may lead to neurodevelopmental disorders.63 Copy number variants of other genes located in the 1q21.1 region, including CHDIL, PRKAG2, and GJA8, also contribute to the risk of both ASD and CHD.59,64
SYNDROMIC AUTISM SPECTRUM DISORDER WITH CONGENITAL HEART DISEASE
The presence of CHD has also been reported in patients with syndromic ASD.11 Various syndromic ASD conditions have shown concurrent CHD, including Fragile X syndrome, Coffin-Siris syndrome, DiGeorge or velocardiofacial syndrome, Phelan–McDermid syndrome, 16p11.2 deletion syndrome, and CHARGE syndrome.53 Fragile X syndrome, primarily caused by FMR1 mutation, exhibits a CHD prevalence of less than 10%.65 This can encompass aortic root dilation, mitral valve prolapses, and arrhythmias.65,66 Coffin–Siris syndrome, a result of loss-of-function mutations in ARID1B, leads to the upregulation of the Wnt/β-catenin signalling pathway.67 This syndrome is associated with a variable degree of neurodevelopmental disability, including ASD.66 Approximately 20% of patients with Coffin–Siris syndrome also present with CHD, which can include mitral valve insufficiency, septal defects, and atrioventricular block.66 DiGeorge, or velocardiofacial syndrome, stemming from the deletion of 22q11.2, has a high CHD prevalence of 75%.68 This may involve conditions such as TOF, pulmonary atresia, truncus arteriosus, and ventricular septal defect.68 Phelan–McDermid Syndrome is associated with terminal deletions of the chromosomal region 22q13.3, which contains the SHANK3 gene.69 CHD is prevalent in this syndrome, with a prevalence of more than 25%.69,70 CHD manifestations can include atrial septal defect, aortic arch anomalies, tricuspid valve regurgitation, patent ductus arteriosus, and total anomalous pulmonary return.69,70 16p11.2 deletion syndrome has a CHD prevalence of 33%, including conditions such as TOF and associated anomalies, and pulmonary atresia.66,71 CHARGE syndrome, attributed to genetic mutations in the CHD7 gene, has ASD prevalence of 15–50%, and a high CHD prevalence of 75–80%.72,73 CHD presentations can include ventricular septal defect, atrioventricular canal defect, TOF, and aortic arch anomalies.72
PATHOPHYSIOLOGY OF CONGENITAL HEART DISEASE LEADING TO AUTISM SPECTRUM DISORDER
The risk factors contributing to the development of ASD in patients with CHD encompass various elements. These risk factors include patient-specific factors, prenatal and perinatal conditions, brain abnormalities, the timing of surgical intervention, and cognitive impairments.2
De novo mutations in shared genetic loci contribute to the overlap between CHD and ASD.40 However, additional patient-specific risk factors can further increase the likelihood of developing ASD in individuals with CHD.2 Foetal and perinatal conditions play a significant role, with levels and duration of hypoxaemia being a crucial stressor in infants with CHD.74 Studies have shown that foetuses with CHD experience abnormal haemodynamic changes that result in alterations in brain circulation.14 This is evidenced by reduced middle cerebral artery pulsatility index and cerebroplacental ratio, leading to reduced brain volume and delay in brain maturation.14 Shunting of blood in foetuses with CHD occurs at atrial, ductal, or isthmic levels to maintain adequate cerebral blood flow.11 The shunting of blood in CHD leads to significant reductions of blood-flow in the major cerebral vessels.75 Studies also showed an increase in cerebral resistance indices using Doppler ultrasound, due to the alterations of blood flow caused by the cardiac defect.76 Furthermore, disruption in pathways that deliver oxygen and substrate-rich blood to the brain, due to intrinsic autoregulatory mechanisms, may lead to an increase in oxygen delivery to the brain to meet the cerebral energy requirements.15 Therefore, the development of ASD in CHD may be due to the burden of pulmonary and circulatory compromise, resulting from brain-sparing circulatory shift changes.77 Additionally, the time to corrective surgery is also an important determining factor, as more injury to the brain occurs when there is a longer wait to the corrective cardiac surgery in CHD infants.78 Longer wait to surgery results in higher extent and duration of cerebral desaturation, which may lead to poor neurodevelopmental outcomes.11
Infants with CHD have a high incidence of brain abnormalities, even with an absence of genetic syndrome.79,80 These abnormalities may arise from abnormal brain development in utero, or from brain injury after delivery.79,80 MRI studies of newborns with CHD showed reduced brain volume for gestational age, decreased metabolism, and delayed cortical folding and development compared to healthy newborns.81-83 Besides that, MRI brain of full term infants also revealed similar findings to preterm infants, including diffuse white-matter injury and focal injury of periventricular leukomalacia, suggestive of hypoxic-ischaemic encephalopathy.8,84
Furthermore, term infants with cyanotic CHD demonstrated similar white matter anomalies and delays in myelination, which are observed in preterm infants.82 Diffusion tensor imaging MRI of infants with CHD has shown a decrease in the white-matter fractional anisotropy and ratio of N-acetylaspartate to choline, which is indicative of disorganisation of white matter structure.79 Damage to the oligodendrocyte progenitor cells and subplate neurons in CHD infants results in impairment of the myelination process and development of white matter track, potentially affecting neural connectivity.80 These structural anomalies can result in emotional processing and social impairments in ASD, associated with atypical cortical and sub-cortical structures, such as the medial prefrontal cortex, orbitofrontal cortex, the right temporo-parietal junction, and amygdala.85 Furthermore, children with ASD have an estimated prevalence of 12–26% of epilepsy.86,87 This increased risk of epilepsy in CHD may be attributed to cerebral hypoperfusion and brain injury during in utero development, and perioperative corrective cardiac surgery.88 Early onsets of seizures after corrective cardiac surgery are associated with poor neurodevelopmental outcomes.89 Therefore, development of epilepsy syndrome in children with CHD may increase the risk of development of ASD.79
Moreover, infants with CHD have a higher rates of adverse perinatal outcomes, including preterm delivery, low birth weight, and perinatal or neonatal infection, all of which can contribute to the risk of developing ASD.90,91 There are two distinct chronological risk factors for the increased risk of ASD in children with CHD, which are during birth and early childhood.92 Furthermore, children and adolescents with CHD often display some extent of social cognition impairments, even if they do not fulfil the diagnostic criteria for ASD.76 For instance, a study by Calderon et al.76 reported that CHD patients who underwent open-heart corrective surgery displayed significant impairments in complex affective mental state understanding, processing social cues, and theory of mind. Notably, the risk of ASD appears to be higher in children with milder forms of CHD, such as atrial septal defects and ventricular septal defects, possibly due to the limited number of cases among children with severe forms of CHD.88
CLINICAL APPLICATIONS, LIMITATIONS AND FUTURE DIRECTIONS
Understanding the genetic links between CHD and ASD may facilitate the comprehensive care and management strategies for affected children. Around 30% of children diagnosed with CHD exhibit syndromic phenotypes, wich include extracardiac manifestations.93 Shikany et al.94 found that 26% of the infants with critical CHD had abnormal genetics test. More widespread genetic testing of patients with CHD, especially in the presence of syndromic phenotypes, should be advocated to identify presence of neurodevelopmental disabilities such as ASD, or extracardiac malformations.95 However, genetic testing in children with isolated CHD may have limited clinical utility due to the high heterogeneity of genetic panels associated with CHD.95 On the other hand, the neurodevelopmental specialist who diagnoses the children with ASD should refer them to a genetic service to identify the presence of underlying medical conditions, single gene defects, or cytogenetic abnormalities.96 However, research shows that only 3% of patients diagnosed with ASD undergo suggested clinical genetic testing, underscoring the discrepancy between expert recommendations and clinical practice.97 Therefore, cardiologists and neurodevelopmental paediatricians should refer the patients with co-occurrence of ASD and CHD for genetic testing and counselling to identify potential underlying genetic mutations.
The existing studies confront several limitations. CHD encompasses a spectrum of severity, ranging from mild left-to-right shunts, such as atrial septal defects, to severe right-to-left shunts such as TOF. Consequently, the risk of ASD may vary depending on the severity of the underlying CHD. Current studies include patients with diverse types of CHD, which might not accurately reflect the true risk of ASD. Moreover, both CHD and ASD result from multifactorial causes, complicating efforts to identify specific genetic mutations implicated in both conditions. Furthermore, the predominant reliance on animal models in genetic studies investigating the co-occurrence of CHD and ASD poses challenges, as the findings may not necessarily translate to human populations. Given the current limitations in our understanding of the neurobiology of ASD, and its obscure pathophysiological mechanisms, elucidating causal relationships between these two conditions presents a formidable challenge. While genetic syndromes provide valuable insights into specific genetic pathways implicated in both ASD and CHD, it may not generalise to non-syndrome cases, due to the phenotypic variability and variable penetrance of associated genetic mutations of the syndromes.
Future research into the genetic associations between CHD and ASD may further improve our understanding of the pathophysiology underpinning both conditions, and inform clinical practice. Firstly, investigating the functional consequences of genetic variants via multi-omics approaches, such as transcriptomics and proteomics, may improve the genetic models of CHD and ASD comorbidity by molecular phenotype profiling.98 Advanced technologies, such as single-cell genomics and spatial transcriptomics, enable high-resolution profiling of cellular heterogeneity and spatial organisation of tissues in children with CHD and ASD comorbidity, thereby improving our understanding of pathogenesis, and allowing the identification of novel therapeutic strategies.99,100 Studies focusing on the gene-environment interactions of CHD and ASD comorbidity will provide insights into the influence of phenotype expression on disease susceptibility.101,102 Furthermore, studies exploring polygenic risk scores and genetic modifiers will be essential to elucidate the complex genetic architecture underlying disease heterogeneity and susceptibility.103
CONCLUSION
There is an increased risk of development of ASD in children with CHD. Therefore, children with CHD should undergo regular follow-up to assess neurodevelopmental outcomes, and screen for ASD symptoms for early diagnosis and intervention.1 Furthermore, family members of children with CHD should be counselled regarding the increased risk of ASD and other developmental disabilities. Further research on the genetics and pathophysiology underlying the co-occurrence of ASD and CHD may aid in understanding the aetiology of the disease, and contribute to the development of novel treatments. Future research should also focus on different prognostic factors, such as genetics, brain abnormalities, haemodynamic changes, and time to surgery, to better elucidate the risk of ASD in children with CHD. This will contribute to the development of more precise risk assessment, and personalised intervention strategies.