Meeting Summary
Prof Fernando Perez-Ruiz opened the symposium, which focussed on the current unmet need in controlling gout and associated comorbidities with current standard of care. Prof Till Uhlig reviewed the epidemiology and pathogenesis of the disease and Prof Thomas Bardin discussed the burden of gout and its comorbidities. Prof Pascal Richette examined the European League Against Rheumatism (EULAR) treatment guidelines and best clinical practices in treating the disease. Prof Alexander So expanded on why current treatment strategies for gout are not reaching satisfactory disease outcomes. Prof Perez-Ruiz and Prof Bardin focussed on dual therapy with new-in-class uricosuric lesinurad, explaining its mode of action and the associated clinical studies, respectively.
Session One: Do You Think You Know Gout? Time to Rethink. Epidemiology and Pathogenesis: A Common Disease with Genetic Predisposition
Professor Till Uhlig
Gout is the most common form of inflammatory arthritis1 and is more frequent than rheumatoid arthritis and spondyloarthropathies, such as psoriatic arthritis and ankylosing spondylitis.2-4 The disease is most prevalent in the USA, with a prevalence rate of 3.9%,2 followed by New Zealand at 2.7%5 and Australia at 1.7%,6 while the rates are lower in Europe,7,8 Mexico,9 and China.10 Gout is predominantly a disease in males with the preponderance in males ranging from 73–82% depending on geographic location.2,5,7,8 Prevalence increases with age for both sexes until ~70 years of age, when it then plateaus at ~7–8% for males and 2–3% for females.11
Gout is primarily a disease related to the purine metabolism, with increases occurring due to a shift in the way purines from internal sources (such as from increased cell turnover, psoriasis, and cancer) or the diet (such as from seafood, meat, beer, and fructose) are eliminated from the body. Purines are metabolised by xanthine oxidase (XO) into uric acid (UA)12 and renal clearance is responsible for ~70% of UA excretion.12 UA is filtered through the kidney’s glomeruli, of which 90% is reabsorbed in the proximal convoluted tubule and 10% is excreted in the urine. The remaining 30% of UA is excreted via the gut.12,13
Reabsorption of UA from the tubule lumen to the bloodstream occurs via transporters on the apical surface of tubular epithelial cells, located in the proximal convoluted tubule.12 URAT1 is the most important transporter involved in UA reabsorption. However, OAT4, OAT10, and Glut9b also function to move UA into the tubular epithelial cells.12,14 Glut9b is known to move UA across the basolateral surface from the tubular epithelial cells into the bloodstream.14
There has been an increase in the number of discoveries of genetic variants associated with hyperuricaemia and gout over recent years,15 with SLC2A9 and ABCG2 exhibiting strong control on serum UA (sUA) levels (Figure 1).16,17 Collectively, SLC2A9 and ABCG2 account for 3.4% of the variance in UA levels, with the SLC2A9 protein acting as a voltage-dependent UA transporter in the kidney and ABCG2 acting indirectly to increase gut excretion of UA.16
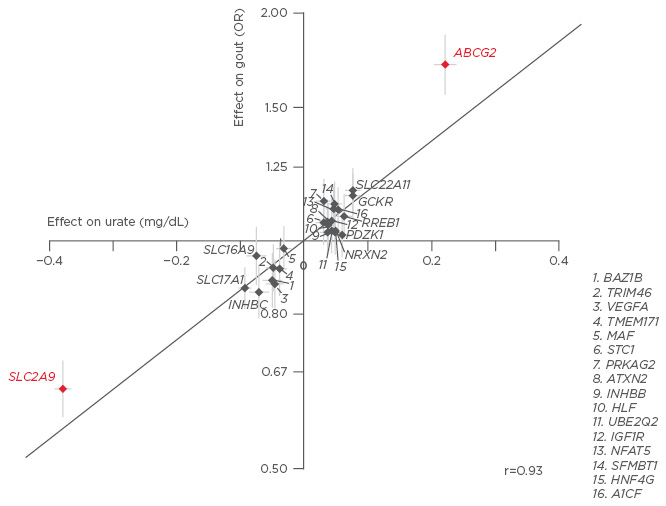
Figure 1: Genetic variants SLC2A9 and ABCG2 exert control on serum uric acid, accounting for 3.4% of the variance in UA levels.16,17
UA: uric acid; OR: odds ratio.
Inefficient renal excretion of UA is the predominant cause of hyperuricaemia (80–90%), whereas the overproduction of UA accounts for the remaining 10–20% of cases.12,18 The solubility threshold of UA has, in one publication, been found to be 6.8 mg/dL (404 µmol/L); above this level precipitation occurs, leading to crystallised UA.18 UA crystal deposition leads to crystal shedding, which activates the inflammatory cascade, resulting in gout flare.19
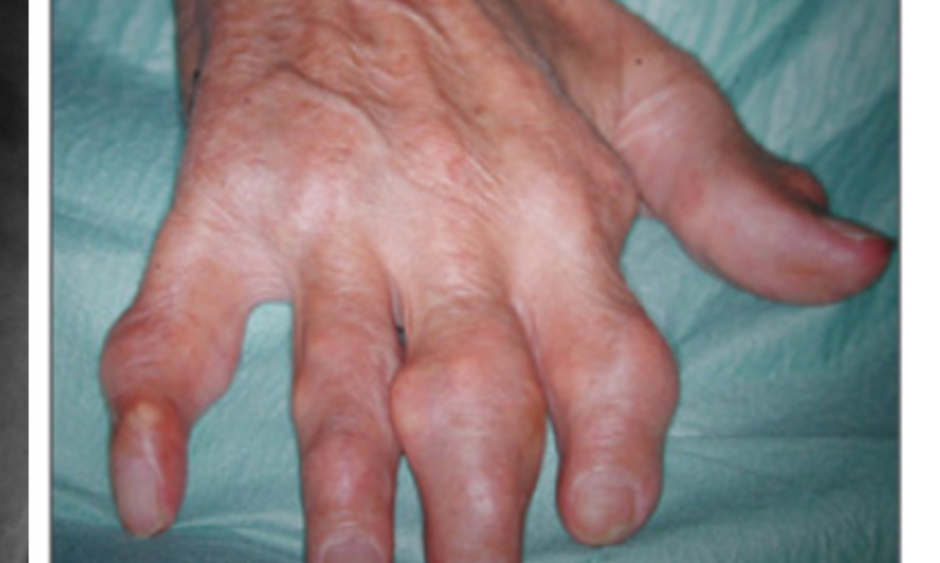
The incidence of a first gout flare increases with rising sUA levels; sUA levels of ≤7.0 mg/dL reflected, in one study, an annual incidence of 0.1%, increasing to 4.9% for very high sUA levels of ≥9.0 mg/dL.20 When left untreated over the course of a year, high sUA levels increase the risk of recurrent gout flares to ~65% for a sUA level of 7.5–8.5 mg/dL and to >80% for sUA levels of >9.0 mg/dL.21 If sUA is left untreated from the initial flare for 1–5 years, 29% of patients will exhibit tophaceous gout. This proportion increases to 47% of patients 6–10 years post initial flare, 61% for 11–15 years post initial flare, and 71% for patients who remain untreated >16 years after the initial flare.22 With increases in tophus sites, there is a noticeable change in the severity of tophi exhibited, with extensive tophi accounting for 2% of the 1–5 year cohort, rising to 13% and 24% in the 16–20 and >20 year cohorts, respectively.22
In summary, gout is a very frequent condition caused by elevated sUA levels, often as a result of inefficient renal excretion of UA.1,12 There are specific gene variants associated with hyperuricaemia and gout.15 If left untreated, high sUA levels lead to crystal formation, which increases the severity of gout symptoms and initiates the inflammatory cascade responsible for gout flare.18,19,23 If such manifestations remain untreated, gout severity will increase over time, along with the likelihood of developing tophaceous gout (Figure 2).22
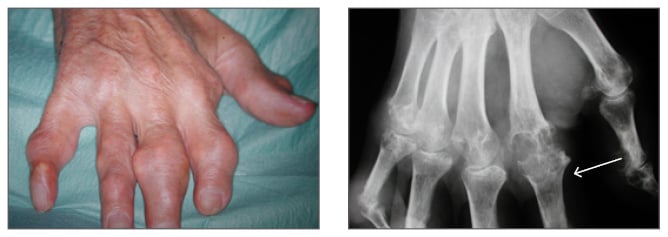
Figure 2: Tophaceous gout of the hand, with X-ray showing bone erosion of index finger (white arrow).
Images courtesy of Prof Fernando Perez-Ruiz, Cruces University Hospital, Baracaldo, Spain.
Disease Burden and Comorbidities: A Chronic Disease with Serious Consequences
Professor Thomas Bardin
Gout is a chronic and progressive condition and is often misperceived as an acute disease by both doctors and patients. As such, doctors focus on treating the symptoms and neglect the hyperuricaemia that leads to crystal deposition. The result is poor disease management; left untreated, elevated sUA levels allow for continued crystal growth, development of tophaceous gout (which can have serious articular consequences), and associated comorbidities.24
In a cross-sectional multicentre study, a cluster analysis of comorbidities and their associations with gout showed that a high proportion of patients with gout had hypertension, Type 2 diabetes mellitus, and metabolic syndrome. The analysis showed that the occurrence of comorbidities increases over time in patients with gout, underscoring the need to treat gout early before the onset of these conditions.25 In a meta-analysis of 17 studies, gout was independently associated with Stage ≥3 chronic kidney disease in 24% of patients26 and was found to increase the risk of end-stage renal disease.27 Gout is also associated with all components of the metabolic syndrome, such as adiposity, hyperuricaemia, and hypertension, although the relationship between each component differs.28 Gout increases the incidence of hypertension and Type 2 diabetes mellitus, and the reverse is also shown to be true, although it is unclear if therapeutic intervention for such comorbidities will lower the risk of gout. However, it is clear that gout is a risk factor for cardiac diseases, such as coronary heart disease (hazard ratio [HR]: 1.26; 95% confidence interval [CI]: 1.14–1.4029 and HR: 1.59; 95% CI: 1.04–2.41),30 heart failure (HR: 1.74; 95% CI: 1.03–2.03),31 and atrial fibrillation.32 In another analysis, gout was found to be associated with a standardised mortality ratio of 2.37 (95% CI: 1.82–3.03), showing an association with premature death for patients exhibiting a high sUA level and tophi.33
Treating patients for elevated sUA levels resolves the underlying cause of disease, enabling monosodium urate (MSU) crystal dissolution and ultimately, cure, although it is worth noting that treatment is required throughout the patient’s life. Once MSU deposits form and begin to grow, the disease becomes increasingly severe and difficult to treat. Severe gout with multiple flares and tophi is classified as a chronic disease, which has a significant impact on patient quality of life, even more so than rheumatoid arthritis.34
In conclusion, gout is a frequently occurring disease that is often misclassified as acute rather than chronic and frequently goes untreated. UA-lowering drugs are known to cure the disease and early intervention is critical. Left untreated, gout can become very severe, with a high rate of cardiovascular and renal complications and increased risk of mortality.
Session Two: Treating to Target? Challenges Galore. Current Treatment Guidelines and Clinical Practice
Professor Pascal Richette
The most effective approach to treating gout is to lower the sUA level to <6.0 mg/dL (<297 µmol/L), or <5.0 mg/dL in severe gout, in order to dissolve crystals in synovial fluid and affected joints,35 and prevent new crystal formation.36 Furthermore, long-term urate-lowering treatment (ULT) has been shown to nearly completely eliminate gout flares in the FOCUS study; the 116 patients in the study did not experience flares requiring treatment after 5 years of febuxostat treatment.37 See Figure 3.38
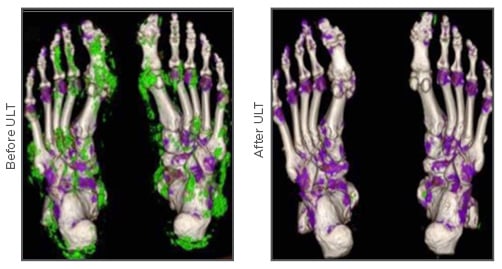
Figure 3: Dual energy computed tomography imaging showing resolution of monosodium urate deposits (green) before and after uric acid-lowering treatment in a patient with tophaceous gout.38
ULT: urate-lowering treatment.
Tophaceous gout can severely impact patient quality of life, and ULT has been shown to significantly lower tophus diameter.18 In a 5-year study of 63 patients with tophaceous gout receiving treatment with allopurinol, benzbromarone, or both, a clear reduction in tophus size was shown, with the largest reductions in tophi found in patients with the lowest sUA levels (1.5 mm per month in patients with a sUA level of <4.0 mg/dL versus 0.53 mm per month with a sUA level of 6.1–7.0 mg/dL).33
The EULAR 2016 guidelines recommend available therapeutic options to decrease sUA levels.39 The first-line of ULT drugs are XO inhibitors (XOI), such as allopurinol, which block XO from metabolising hypoxanthine to xanthine, and then xanthine to UA and thus decrease the production of UA, thereby reducing overall levels of sUA.12 Uricosurics, such as probenecid, sulfinpyrazone, benzbromarone, and, more recently, lesinurad, act on the rate of UA reabsorption in the kidney;13 they are prescribed as second-line treatment when an XOI alone is inadequate in lowering sUA or not tolerated by the patient.39
The EULAR guidelines recommend that patients be treated from first presentation of the disease, especially patients presenting at a young age, high sUA levels, or with multiple comorbidities.39 Treatment should be initiated with a low dose of allopurinol (50–100 mg per day),40 titrated by 100 mg increments every 2–4 weeks up to 800–90041,42 mg per day if appropriate, to reach the urate target (6.0 mg/dL). For patients with severe gout, a sUA level of <5.0 mg/dL is recommended for faster tophi dissolution.39 Titration aims to decrease the risk of flare and allergic reaction.40 The allopurinol dose should be adjusted for renal function based on creatinine clearance.43 Patients who do not respond to allopurinol should be switched to febuxostat, or a combination regimen of XOI/uricosuric treatment. The guidelines also stress the importance of patient education in areas such as lifestyle (weight loss and diet) in the effective management of flares and long-term reduction of sUA levels. Systematic screening for comorbidities, such as renal impairment and cardiovascular disease, is part of gout management.
Primary care studies have shown that treatment is poorly concordant with guidelines, as evidenced by a lack of patient education, restriction of allopurinol to a minority of patients, inappropriate dosing, lack of sUA level monitoring, and suboptimal screening for comorbidities.44-47
Overall, the goal of ULT is to maintain low enough sUA levels to prevent MSU crystal formation and disposition and to promote dissolution. Guidelines recommend that treatment with ULT should decrease sUA levels to <6.0 mg/dL and that patients are educated about their disease and how they can modify their lifestyle.
Unmet Medical Needs and Limitations of Current Treatments
Professor Alexander So
Effective treatment of gout means lowering sUA to below the target level (<6.0 mg/dL). Although diet influences the risk of developing gout48 and dietary change is often recommended to reduce the risk of incident gout, the effect of diet on established hyperuricemia is small. The maximum reduction in sUA level achieved with dietary intervention alone is only ~1 mg/dL, which is not sufficient to reach the desired treatment target.18 Furthermore, there is little evidence that dietary education leads to significant change in serum urate in gout patients.49
Drug treatment is the mainstay of effective urate lowering. XO inhibition is the main mode of action of current ULT. With 95% of the market share, allopurinol is the most widely used XOI12 due to its availability, once-daily dosing regimen, efficacy, and low cost.18 Failure to meet desired sUA endpoints with allopurinol ULT is frequently because the dose is too low to enable the patient to reach target sUA levels, as was shown in the FACT,50 APEX,51 and CONFIRMS52 trials. In these trials, 58–61% of the patients who were prescribed 100–300 mg allopurinol did not achieve an sUA level of <6.0 mg/dL by their final visit. In LASSO,53 an open-label, uncontrolled, multicentre, 6-month study of 1,732 gout patients, only 19% of patents were treated with >300 mg allopurinol per day and even at a dose >300 mg only 45.9% of patients achieved target sUA levels.
The majority of patients with gout are under-excretors rather than over-producers of UA.12,18,42,54 These patients may achieve lower sUA levels when prescribed uricosurics, which influence renal excretion of UA.12
Currently, there is a significant unmet need for new molecules that act on urate excretion in gout patients. Benzbromarone, probenecid, and sulfinpyrazone are the currently available uricosurics, but each drug has a tolerance profile that has restricted their clinical use. Although the 2016 EULAR guidelines recommend combination therapy with an XOI and a uricosuric when XOI monotherapy is unsuccessful,18 there are only a small number of observational studies that have documented combination therapy’s effectiveness.
Session Three: The Start of a New Era: Lesinurad: A Novel, Selective Uric Acid Reabsorption Inhibitor
Professor Fernando Perez-Ruiz
Lesinurad is a first-in-class UA reabsorption inhibitor that enhances UA excretion by selectively inhibiting reabsorption of UA by URAT1 and diuretic-induced hyperuricemia by OAT4 in the proximal tubule of the kidney.39,55 Compared with other uricosurics, lesinurad exhibits minimal drug–drug interactions and side effects.56
When used in combination with allopurinol, lesinurad targets inefficient renal UA clearance in patients, as shown in the Phase I clinical trial RDEA594-110.56 There are no clinically relevant drug–drug interactions between lesinurad and XOI.57 Lesinurad is approved only for use in combination with XOI and not as a monotherapy.58 Lesinurad is eliminated through urinary excretion with CYP2C9 as the primary means of hepatic metabolism.58 Unlike benzbromarone, lesinurad does not induce mitochondrial hepatotoxicity.59 Lesinurad has a half-life of ~5 hours, with >60% of the dose excreted in urine, of which 30% is not metabolised.60 Although renal excretion is rapid, lesinurad should be used with caution in CYP2C9-poor metabolisers.
Lesinurad Clinical Development Programme
Professor Thomas Bardin
Lesinurad is the only uricosuric to have undergone a complete clinical development programme (Phase I–III), leading to its indication as a combination treatment for patients with gout who have not achieved the sUA target with an XOI treatment alone.61,62 Three pivotal Phase III trials were conducted; CLEAR1 (N=603) and CLEAR2 (N=610)63 evaluated a combination of lesinurad/ allopurinol, and CRYSTAL (N=324) evaluated lesinurad/febuxostat dual treatment. All three trials had a core study period of 12 months and an extension of a further 12 months.62,64,65
Inclusion criteria for patients with gout in the CLEAR studies included an initial sUA level of ≥6.5 mg/dL (387 μmol/L), being on a stable dose of allopurinol between 300 mg and 800/900 mg (200 mg acceptable for patients with renal impairment) for a minimum of 8 weeks prior to study entry, and two or more gout flares in the previous 12 months; there was no exclusion for a history of kidney stones. The primary endpoint was sUA <6.0 mg/dL by Month 6, and the secondary endpoints were mean flare rate from Month 6 to 12, and complete resolution (CR) of tophi.66 Patients were randomised into three arms (200 mg lesinurad + allopurinol, 400 mg lesinurad + allopurinol, or placebo + allopurinol). Colchicine or non-steroidal anti-inflammatory drugs (NSAID) were given for gout flare prophylaxis from 2 weeks prior to the start of the trial until Month 5. Patients were predominantly male (94.0–96.2%) with an average age of 51.6 years, and an average BMI of 34.5. Patients had been diagnosed with gout for an average of 11.65 years, with a baseline sUA level of 6.9 mg/dL. Several patients (9.0–15.9%) exhibited tophi across an average of 1.9–2.2 sites. Around 65%, 45%, and 15% of patients had comorbid hypertension, hyperlipidaemia, and Type 2 diabetes mellitus, respectively.63,66
Significantly greater (~2.0–2.5 fold) proportions of patients achieved the target sUA level of <6.0 mg/dL at Month 6 with lesinurad (400 mg or 200 mg) and allopurinol compared with allopurinol alone in both CLEAR1 (59.2%, 54.2%, and 27.9%, respectively) and CLEAR2 (66.5%, 55.4%, and 23.3%, respectively) trials. This target was consistently met over the 12-month and 24-month extension studies for both lesinurad doses.66 The proportion of patients with CR of at least one target tophus continued to increase over time, from 10–15% of patients at Month 6 to 34–44% of patients at ≥18 months.67 Finally, the patients receiving lesinurad with allopurinol in the initial study showed a continued decline in gout flare that required treatment throughout the extension period.67
The CRYSTAL study included patients with tophaceous gout and sUA level of >8 mg/dL (not on ULT) or >6.0 mg/dL (if on ULT). Patients were treated with lesinurad 200 or 400 mg/day in combination with febuxostat 80 mg/day and colchicine or NSAID for gout flare prophylaxis from 2 weeks prior to the start of the trial and continued up to Month 5; the primary endpoint was sUA <5.0 mg/dL by Month 6, and key secondary endpoint at Month 12 of either CR or partial resolution of tophus. Patients were predominantly male (95.4%), with a mean age of 54.1 years±11.0 standard deviation (SD). Average sUA at screening was 8.7 mg/dL±1.6 SD, with 1.8±1.2 SD tophi and 6.7±8.2 SD gout flares in the preceding 12 months.62
Lesinurad 400mg and 200mg in combination with febuxostat resulted in sustained reductions in sUA to <5.0 mg/dL at Month 6 in 76.1% and 56.5% of patients, respectively,compared to febuxostat monotherapy in 46.8%. The mean sUA level remained at ≤~3.5 mg/dL for the entire 12-month study. There was a trend of continued decrease in tophus size across all study arms, with significant observations at 12 months for both lesinurad/febuxostat groups where the mean change was a 55.8–57.9% (p<0.05) decrease with a significant reduction in total target tophus area from baseline versus febuxostat alone.62 Overall, the proportion of subjects with CR of at least one target tophus continued to increase over time; the proportion of patients with fewer gout flares requiring treatment also increased across all treatment groups in the extension study.
The pooled safety data for lesinurad use in CLEAR1, CLEAR2, and CRYSTAL were critically evaluated, as these studies are the first to validate the combinatory use of lesinurad and XOI. Due to lesinurad’s mode of action as a uricosuric drug, renal tolerance was closely assessed. XOI monotherapy revealed increased serum creatinine (sCr) in 2.3% of patients, which was elevated to 4.3% in the lesinurad 200 mg/XOI group and to 7.8% in the lesinurad 400 mg/XOI group. However, these elevations were both transient and reversible, with most cases of sCr elevation resolved by the time of the next assessment despite the drugs continuing to be administered.66 Additionally, low rates of kidney stones were reported in patients throughout all arms of the CLEAR studies (<3%), especially for the 200 mg lesinurad/allopurinol cohort (0–1%).63
In conclusion, lesinurad is the only uricosuric to date that has randomised controlled trial evidence and underwent a complete clinical development programme. It is a selective uricosuric and, in combination with a XOI, led to greater improvement in sUA levels, tophi resolution, and rate of gout flares than XOI alone.61,62,66 Overall, lesinurad in combination with an XOI was generally well tolerated;68 lesinurad is associated with higher rates of sCr elevation compared with XOI alone, and a low rate of kidney stone treatment-emergent adverse effects.66 For these reasons, lesinurad should be considered an appealing option for use in addition to XOI in patients who fail to achieve sUA targets with XOI therapy alone.