Meeting Summary
This symposium was held during the 2023 European Respiratory Society (ERS) International Congress in Milan, Italy. The main objective was to discuss unmet needs in the diagnosis and management of rare lung diseases, with a particular emphasis on alpha 1 antitrypsin deficiency (AATD) and idiopathic pulmonary fibrosis (IPF). Presentations focused on promising approaches to meet these needs, including the feasibility of genomic diagnosis, the development of improved biomarkers of disease progression, such as quantitative CT and novel blood biomarkers, the use of digital lung auscultation, and increased screening for AATD in vulnerable populations.
The overarching message from the symposium was that advancements in technology, multidisciplinary collaboration, and partnerships between academic institutions, patient associations, and industry are crucial to the continued improvement of patient management in rare diseases, and that the education of healthcare professionals is vital to enhance the understanding and awareness of these conditions.
Identifying Unmet Needs in Rare Lung Diseases
Luca Richeldi
Luca Richeldi, Professor of Respiratory Medicine at the Catholic University of the Sacred Heart and Head of Pulmonology at the A. Gemelli Hospital in Rome, Italy, explained that rare diseases represent a worldwide healthcare challenge, with approximately 10,000 disorders affecting 6% of the population in Western societies.1 Over 80% of rare diseases have a genetic component, which makes these conditions particularly disabling and expensive to manage in terms of genetic testing and comprehensive management.1
Most patients with a rare disease do not receive a correct diagnosis after standard diagnostic testing, and one-third of children with a rare disease die before their fifth birthday, presenting a clear unmet medical need.1
As an example of the obstacles associated with identifying a rare disease, Richeldi described the vast number of conditions that can cause bronchiectasis, including three congenital disorders: cystic fibrosis, primary ciliary dyskinesia, and AATD.2 Richeldi explained that the identification of a condition causing bronchiectasis can require a multitude of different diagnostic tools, such as sweat chloride testing and CFTR mutational analysis if cystic fibrosis is suspected, and alpha 1 antitrypsin (AAT) levels and phenotyping if AATD is suspected.2
Richeldi stressed that research into rare lung diseases often has value beyond the rare disease itself.3-5 For example, the pathogenesis of AATD can be seen as a paradigm of conformational diseases,3 and research into IPF has led to a greater understanding of the spectrum of pulmonary fibrosis.4,5
Richeldi emphasised that many rare diseases have a genetic component, and recent research in the UK and Ireland has indicated that genomic diagnosis of rare paediatric disease is feasible.6 However, this same study showed that the diagnosis of these disorders is significantly less likely in patients of African descent than those of European descent (odds ratio [OR]: 0.51).6
In conclusion, Richeldi identified five unmet needs in rare disease: a lack of precise and widely available diagnostic tools; a lack of prognostic tools; the challenges in designing, performing, interpreting, and funding randomised clinical trials; the heterogeneity in rare disease, which makes it difficult to target them as a single entity; and a high risk of disparities in diagnosis and treatment for ethnic and socio-economic minorities.
Challenges and Progress in Predicting Disease Trajectories
Noel G. McElvaney
Alpha 1 Antitrypsin Deficiency
AATD is a hereditary condition that predisposes patients to early-onset chronic obstructive pulmonary disease (COPD), liver cirrhosis, neutrophilic panniculitis, and systemic vasculitis.7 The AAT gene, SERPINA1, has two alleles, with most individuals carrying the Pi*MM genotype (where Pi* is an alias for SERPINA1). Patients with AATD generally present with genotypes Pi*MS, Pi*SS, Pi*MZ, Pi*SZ, or Pi*ZZ, and the majority (96%) of individuals with AATD-associated disease exhibit the Pi*ZZ allele in clinical practice.7
Noel G. McElvaney, Head of the School of Medicine at the Royal College of Surgeons in Ireland (RCSI), University of Medicine and Health Sciences in Dublin, Ireland, presented data from the long-term follow-up of Irish patients included in the RAPID and RAPID extension (RAPID-OLE) studies. The RAPID study was a multicentre, randomised, double-blind trial comparing the efficacy and safety of AAT treatment with placebo over 24 months (N=180).8 At the end of the RAPID study, 140 patients entered the open-label, 24-month RAPID-OLE study.8,9
After 24 months, AAT treatment was associated with a significantly lower reduction from baseline in lung density at total lung capacity compared with placebo (-1.51 g/L/year versus -2.26 g/L/year [p=0.021]).9 Those patients remaining on AAT treatment in the RAPID-OLE study continued to show a similar rate of lung density decline over the following 24 months (-1.63 g/L/year), whereas those patients who switched from placebo to AAT showed a showed a significantly reduced rate of decline (-1.26 g/L/year) compared with prior placebo treatment (mean treatment difference: 0.52 g/L/year; p=0.008).9
McElvaney explained that since the few patients who reached a terminal event during the RAPID study did so at a lung density of approximately 20 g/L, delaying the decline in lung density with AAT treatment could potentially extend survival in patients with AATD by approximately 5–6 years.9
To investigate this hypothesis, a study is being conducted in Ireland to follow up patients with AATD over a period of 10 years.10 The treatment cohort includes patients with AATD (Pi*ZZ) who participated in the RAPID/RAPID-OLE studies (n=19), and who have continued AAT therapy for up to 10 years after study cessation. Matched controls include patients with the Pi*ZZ genotype (n=19) who have not received AAT, and patients with the Pi*MM genotype (n=20) who had a similar disease severity to the treatment group at baseline.10
Interim results from this study indicate that spirometry, quality of life, and the diffusing capacity of the lungs for carbon monoxide are of limited use in AATD to detect progression over time. These findings are in line with an analysis of long-term registry data from Ireland, Switzerland, and Austria, in which 431 lung index cases and 178 non-lung index cases showed a plateau in forced expiratory volume decline in their late 40s.11 That study reported a significantly lower mortality during follow-up time among those who received AAT therapy compared with those who did not (p=0.00076; Figure 1).
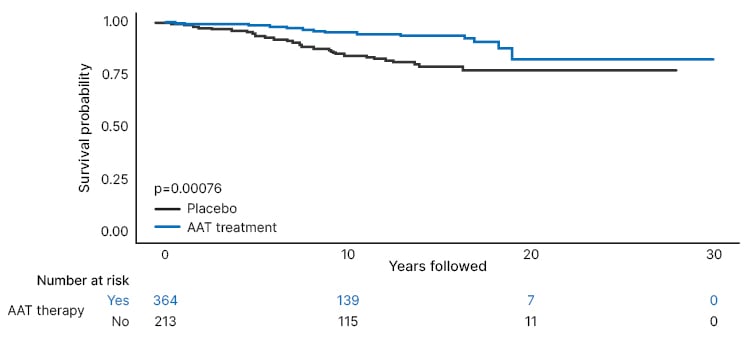
Figure 1: Kaplan–Meier survival analysis by alpha 1 antitrypsin therapy status.
Reproduced with permission from Fraughen et al.11
AAT: alpha 1 antitrypsin.
McElvaney concluded that lung function parameters are not informative to predict disease progression, and are largely decoupled from mortality.10,11 However, it is possible that imaging biomarkers such as high-resolution CT (HRCT) lung density will be better able to monitor disease progression in patients with AATD, and may also provide further insights into an individual patient’s prognosis.
McElvaney explained that healthcare systems may need to extend the diagnostic reach for AATD beyond White, European populations, to produce diagnostic algorithms for general practitioners, to consider neonatal screening, and to increase access to treatments, such as plasma-purified AAT, around the world.
McElvaney emphasised that AATD may not necessarily be a truly rare condition, but one that is diagnosed rarely. Finally, McElvaney stressed that AATD is probably the best clue that clinicians have for what causes COPD, and that a better understanding of AATD will lead to a better understanding of the pathophysiology and the mechanistic causes of COPD.
Idiopathic Pulmonary Fibrosis
Marlies S. Wijsenbeek
IPF is a rare interstitial lung disease (ILD) characterised by worsening dyspnoea and progressive loss of lung function, which arises from progressive scar formation in the lungs.12
Marlies S. Wijsenbeek, a pulmonologist and Chair of the ILD centre at the Erasmus Medical Centre (MC) in Rotterdam, the Netherlands, explained that patients with IPF experience a significant decline in lung function of approximately 200 mL per year.12 However, the disease course varies considerably, with some patients experiencing a rapid decline in lung function, some a more gradual decline, and some patients experiencing acute exacerbations associated with a high rate of mortality.12,13
Predicting the disease course and prognosis in IPF is important because treatment is intended to slow the decline of progressive disease, and the frequency of follow-up often depends on the rate of lung function decline. Similarly, Wijsenbeek explained that patient prognosis has an impact on lung transplant referral, advanced care planning, and clinical trial inclusion. For the patient with IPF, their prognosis has an impact on how they weigh treatment decisions, make plans for their future, and prioritise different aspects of their lives.
One of the first clinical measures used to predict mortality in IPF was the gender-age-physiology (GAP) index.14 However, since the development of the index, the diagnostic criteria for IPF and the treatments available have advanced, and Wijsenbeek explained that the original GAP score now overestimates mortality. Several novel clinical predictors of survival have been explored, and univariate analysis found that 6-minute walk distance (6MWD) and the presence of exertional hypoxia each outperformed the predictive value of the GAP index and remained independently associated with mortality in the multivariate analysis (hazard ratio [HR]: 2.49 [p<0.001]; HR: 2.92 [p<0.001]; and HR: 1.23 [p=0.022], respectively; N=562).15
Patient-reported outcomes predictive for survival in IPF include change in the Living with Pulmonary Fibrosis (L-PF) questionnaire dyspnoea score16 and change in body weight.17 Events such as a forced vital capacity loss ≥10% per year,18 acute exacerbations,18 and worsening respiratory symptoms requiring hospitalisation have also been shown to be predictive of mortality in patients with IPF.19,20
Wijsenbeek explained that advanced techniques are being developed to support the interpretation, and therefore predictive value, of HRCT scans, such as deep learning-based algorithms.21 In addition, blood biomarkers have been identified that can help to predict progression in IPF, including epithelium-derived biomarkers and monocyte counts,22,23 and proteomic biomarker panels have been developed.24,25 Short telomere length has also been found to be associated with disease progression in both IPF and other forms of progressive pulmonary fibrosis.26
Finally, an experienced clinician’s opinion regarding disease prognosis is of considerable value. In a prospective study of 140 patients, clinicians were asked the ‘surprise’ question: “Would you be surprised if this patient died within the next year?” The study found that a clinician’s response was of significant value in predicting 1-year mortality in patients with IPF (OR: 3.69; p=0.019).27
While advances have been made in predicting mortality, Wijsenbeek emphasised that it remains challenging to predict disease progression for the individual patient with IPF,28 and better tools are needed to address this challenge to guide patient treatment and care.
Camouflage: How Asthma Obscures the Diagnosis of Alpha 1 Antitrypsin Deficiency
Filipa Costa
The symptoms of AATD, such as wheezing, phlegm, and dyspnoea, appear at a young age,29,30 are non-specific, and overlap with more common diseases such as asthma.31,32 Asthma is the most common respiratory misdiagnosis prior to diagnosis of AATD,33 and most patients with AATD are initially treated for asthma.31
Filipa Costa, a respiratory physician at the Coimbra Hospital and University Centre in Portugal, stressed that AATD and asthma are distinct disease entities. While asthma is characterised by reversible airflow obstruction, and inflammation and hyper-reactivity of the airway, AATD is characterised by non-reversible airflow obstruction, and responds poorly to inhaled steroids used in the treatment of asthma.31 However, there remains significant overlap between the pathobiology, symptoms, functional characteristics, and key clinical features of these two conditions,33-36 contributing to misdiagnosis.
In addition, it has been suggested that these two diseases may merge to form an overlapping syndrome.37 Although the prevalence of asthma in patients with AATD is variable, 14% of patients with AATD have been reported to have a concomitant diagnosis of asthma.38,39 The presence of coexisting asthma and AATD has been shown to exacerbate symptoms, and these patients are at increased risk of more severe disease,40 suggesting that AATD may represent a risk factor for both developing asthma and for worsening its course.41
Although the biological mechanisms linking these two conditions are not fully understood,42 both conditions are associated with an excess of neutrophil elastase, suggesting a shared pathway of inflammation.37,43 In addition, AAT has important anti-inflammatory and immunomodulatory effects,37 and reduced AAT levels in patients with AATD could potentially lead to chronic airway inflammation and susceptibility to asthma.31,42
Costa explained that distinguishing AATD from asthma based on presentation and clinical evaluation is not possible due to overlapping clinical features.32 However, improving diagnosis in this population is fundamental to the optimisation of clinical management,32 since AATD is treated with AAT replacement therapy, whereas asthma treatments aim to reduce bronchial hyper-responsiveness and inflammation.37
Current guidelines recommend screening for AATD in all patients with COPD, and adults and adolescents with asthma,44,45 and studies indicate that screening may result in the detection of more patients with AATD at a younger age, allowing for implementation of earlier lifestyle changes and therapy.31,46,47 Screening for AATD requires a simple blood test for serum AAT levels.31,32 An AAT level in the normal range rules out AATD, whereas low AAT levels should prompt phenotyping or genotyping for common mutations (Figure 2).
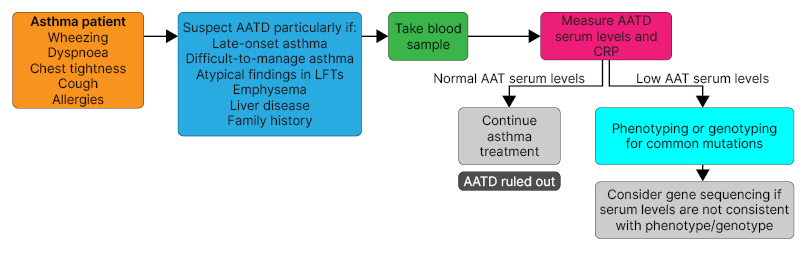
Figure 2: Diagnostic flow diagram for the screening of alpha 1 antitrypsin deficiency in patients
with asthma.
Reproduced with permission from Costa.
AAT: alpha 1 antitrypsin; AATD: alpha 1 antitrypsin deficiency; CRP: C-reactive protein; LFT: lung function test.
However, Costa stressed that despite recommendations, most patients who are candidates for screening are not tested for AATD.31 While screening for AATD should be performed in all adolescents and adults with asthma,44,45 Costa described several disease features that should alert clinicians to a suspicion of AATD, including late-onset and/or difficult-to-manage asthma; atypical findings in lung function tests; evidence of hyperinflation on chest radiographs or emphysema on HRCT; presence of liver disease; and a family history of COPD or liver disease.
Imaging Challenges in Lung Fibrosis
Simon Walsh
Simon Walsh, a thoracic radiologist and National Institute for Health and Care Research (NIHR) Clinician Scientist at the National Heart and Lung Institute, Imperial College London, UK, described three challenges to the management of patients with pulmonary fibrosis: diagnostics, disease behaviour prediction, and early detection, all of which relate to imaging, and all of which are, in principle, amenable to deep learning solutions.48
Challenge 1: Diagnostic Agreement
While guidelines are available for the evaluation of pulmonary fibrosis from HRCT scans,49 visual assessment of fibrosis can be associated with poor reproducibility, interobserver variability, and insensitivity to marginal progression.48,50 For example, interobserver agreement for these histopathologic patterns across 150 HRCTs of patients with pulmonary fibrosis indicated a median κ coefficient of 0.48–0.52 across 112 radiologists50 (where 1.00 is perfect agreement, 0.40 is generally considered to be clinically acceptable, and 0.00 is agreement by chance).
To investigate the potential for computer-based diagnostic support in the setting of pulmonary fibrosis, deep learning was used to develop an algorithm able to categorise histopathologic patterns on HRCT (the Systematic Objective Fibrotic Imaging Analysis Algorithm [SOFIA]).51 Interobserver agreement between the algorithm and consensus radiologist opinion (across 91 thoracic radiologists) was assessed in 150 HRCT scans.51 The individual radiologists’ interobserver agreement with the consensus opinion reached a median κ coefficient of 0.67, and the agreement of the algorithm with the consensus opinion reached a κ of 0.69, showing that the algorithm outperformed radiologists in this study. In addition, the algorithm provided equivalent prognostic discrimination between usual interstitial pneumonia and non-usual interstitial pneumonia patterns to the consensus opinion of the thoracic radiologists.51
Challenge 2: Identification of Progressive Pulmonary Fibrosis
Walsh explained that, once disease is established, it is not currently possible to reliably predict which patients will develop progressive fibrosis and which will remain stable from baseline information alone.48 SOFIA was able to provide probabilities for the occurrence of each of the pulmonary fibrosis diagnostic categories across a large cohort of patients with progressive pulmonary fibrosis. Analysis showed that SOFIA-based probabilities, but not expert radiologist consensus, were predictive of mortality (HR: 1.75; p<0.0001), and that this was independent of disease extent (n=83).52
Challenge 3: Early Detection of Pulmonary Fibrosis
Patients are typically diagnosed when they present with symptoms, yet Walsh emphasised that disease is present well before this occurs. Incidentally detected interstitial lung abnormalities have been associated with IPF,48 yet only a small proportion of patients with these abnormalities go on to develop IPF.48,53,54 The difficulty lies in how to predict progressive pulmonary fibrosis based on subclinical CT abnormalities like these, a challenge that may be amenable to computer-based CT analysis.48
Walsh explained that the scarcity of CT data for progressive pulmonary fibrosis presents a challenge to algorithm training.48 While generative artificial intelligence (AI) and synthetic data can be used to train deep learning algorithms,55 the ideal solution is to collate sufficiently large datasets, such as the vast repository of ILD data that are being gathered by the Open Source Imaging Consortium (OSIC).56
In closing, Walsh reiterated that AI is intended to supplement the expertise of radiologists, rather than replace them, in the manner of automated decision support, with a view to improving the diagnosis and management of pulmonary fibrosis.
New Tools to Advance Imaging in Rare Lung Diseases
Elizabeth Estes
Elizabeth Estes, Executive Director of the OSIC in Saugatuck, Michigan, USA, explained that the mission of OSIC-ILD is to make radical progress on behalf of people living with ILDs, their families, and their caregivers. The OSIC aims to become the largest and most diverse digital image biobank for ILDs and other rare lung diseases in the world, with the intention of driving technological innovation in a clinically meaningful way.56
By developing a large collective of real-world data, the OSIC will support the development of technology to speed diagnosis, and improve prognosis and response to therapy, in ILD and other diseases. The OSIC-ILD repository includes a total of 20,752 committed datasets,56 with 4,924 anonymised patients and 6,322 anonymised CT scans (OSCILD.org, unpublished data).
OSIC’s large repository of CT scans is intended to overcome challenges to the development of deep learning algorithms; these include an insufficient number of available lung CT scans, primarily homogeneous populations, and limited collaboration between study groups. The OSIC repository aims to improve the accuracy of generative AI algorithms and help to remove variability from image analysis.56,57 The OSIC also plans to test the multitude of member algorithms against a similar, locked dataset from the OSIC, to encourage sharing and debate among scientists. The data contained in the OSIC repository will also support the identification of novel biomarkers in ILD.56
The next steps for the OSIC-ILD include increasing the number of longitudinal scans and clinical follow-up of patients with ILD in the database, and adding scans and clinical data from patients with AATD and sarcoidosis.
Estes encouraged all clinicians to consider the way in which data need to be captured in the clinic to maximise the contribution to algorithm-based research, and to support the OSIC to get newly developed and validated tools into the clinic, where they can support patients.
Implications of Unmet Needs in Rare Disease for Management of Rare Lung Diseases
Luca Richeldi
Richeldi reiterated that key unmet needs in rare disease include a lack of diagnostic/predictive tools and a high risk of inequality in diagnosis and treatment. Richeldi stressed that patients with IPF may be asymptomatic, which can make cases challenging to identify,49 and this underlines the need for inexpensive and widely available diagnostic tools that can be used in all parts of the world.
As an example of a widely available diagnostic tool that could help to identify patients with rare disease, Richeldi described the value of lung auscultation in the diagnosis of IPF. Lung auscultation can be used to detect bibasilar inspiratory (‘velcro-type’) crackles, and guidelines indicate that the presence of this clinical sign should lead clinicians to suspect IPF.49,58
The presence of bibasilar inspiratory crackles has been shown to predict the radiologic HRCT features of fibrosis (OR: 6.24), traction bronchiectasis (OR: 4.37), reticulation (OR: 2.57), honeycombing (OR: 2.39), and ground glass opacities (OR: 2.13), each p<0.001.59 In addition, quantitative analysis of lung sounds (producing reproducible, distinct digital acoustic features) showed that the specificity and sensitivity of acoustic features to identify fibrosis of ≥50% on HRCT were similar to that of a composite physiological index, which incorporates physiology and imaging data (area under the curve: 0.73 and 0.69, respectively; Figure 3).60
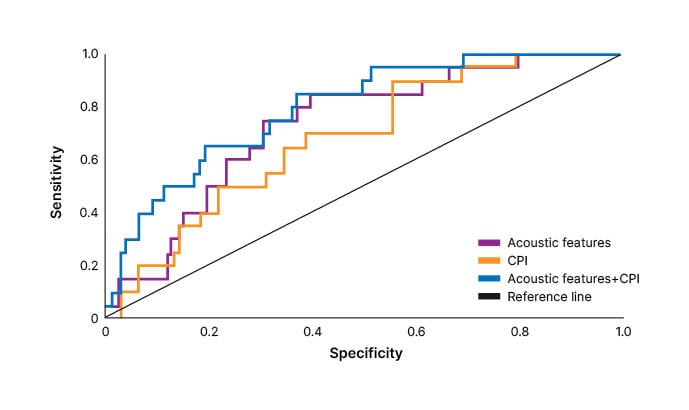
Figure 3: Receiver operating characteristic curves of acoustic features and/or composite physiological index for identification of extent of fibrosis ≥50% on high-resolution CT scans.
Reproduced with permission from Sgalla et al.60
CPI: composite physiological index.
These studies indicate that digital lung auscultation may be relevant for the screening and early detection of patients with IPF and other forms of pulmonary fibrosis, and that it may represent a cost-effective way to monitor disease progression.61
Richeldi explained that advancements in technology and multidisciplinary collaboration will undoubtedly help to address unmet needs in rare disease. Partnership between academic institutions, patient associations, and industry is also crucial, but can pose a challenge because of the different agendas and driving forces among these groups. Finally, Richeldi stressed that education remains the most critical factor in addressing the unmet needs in rare disease.
Summary and Closing Remarks
The main improvements needed in the field of rare lung diseases relate to diagnostic and prognostic tools, disparities in diagnosis and treatment for minority populations, education of the medical community, and collaboration within multidisciplinary teams.
The symposium presenters discussed promising developments in the evaluation of lung disease, such as novel biomarkers; short telomere length; digital lung auscultation; patient-reported outcomes; and reproducible, quantitative HRCT scan measures, which can take advantage of deep learning-based automated decision support. The OSIC may be of benefit in this regard since it provides an umbrella group to standardise HRCT scans. It was also noted that while forced expiratory volume decline tends to plateau in patients with AATD, AAT therapy continues to provide a survival advantage in this population.
The presenters also emphasised the importance of screening for rare lung disease, such as screening for AATD in with adult-onset asthma, and the potential for genomic screening in the diagnosis of paediatric rare diseases in the future.






