Meeting Summary
This symposium took place during the 2023 meeting of the European Respiratory Society (ERS), with a focus on targeting chronic obstructive pulmonary disease (COPD) with Type 2 inflammation, and the emerging biologic landscape. The speakers discussed the clinical consequences of COPD through an understanding of pathological changes, the spectrum of inflammatory pathways, the role of Type 2 inflammation in the pathophysiology of COPD, and the evolving clinical landscape in COPD. Klaus Rabe, Full Member (Chair), LungenClinic, Grosshansdorf, Germany, utilised hypothetical clinical scenarios to contextualise the clinical presentation of COPD as a consequence of disease pathology, specifically chronic inflammation leading to structural changes of airways and parenchymal destruction resulting in airflow limitation, leading to worsening symptoms, and increasing further exacerbation risk. Stephanie Christenson, Assistant Professor of pulmonology at the University of California, San Francisco, USA, followed with a discussion of the heterogeneity of inflammatory pathways, exploration of distinct inflammatory cells and cytokines, and the evolving state of the knowledge of the diverse inflammatory pathways associated with COPD. COPD inflammation can be differentiated by distinct inflammatory cells and cytokines into Type 1/Type 3 inflammation (i.e., neutrophilic inflammation) and Type 2 inflammation. However, there is potential overlap in the various inflammatory mechanisms driving COPD via the alarmins IL-33 and thymic stromal lymphopoietin. In addition, the key cytokines IL-4, IL-13, and IL-5 mediate the pathophysiology of COPD with Type 2 inflammation. Altogether, the heterogeneous inflammatory pathways contribute to characteristic features of COPD, fibrosis (small airways), wall thickening, airway remodelling, and clinical features, such as shortness of breath at rest. Dave Singh, Professor of respiratory pharmacology at The University of Manchester, UK, then discussed active areas of investigation in the development of additional treatments for patients with COPD.
Contextualising Clinical Presentation of Chronic Obstructive Pulmonary Disease Through Disease Pathology
Klaus Rabe
Rabe began the presentation by highlighting two hypothetical patients with different ages, physiologies, and backgrounds to contextualise the clinical presentation of COPD.
Leonard is a 52-year-old patient with COPD, who is uncontrolled, on double therapy (long-acting β-agonist [LABA] or long-acting muscarinic antagonist [LAMA]), and for whom inhaled corticosteroids are contraindicated. His current symptoms include shortness of breath going uphill or taking stairs, constant cough with mucus production, and disruptive anxiety about his future. His BMI is 32 kg/m2, and he is a former smoker with a 15 pack-year history. His laboratory assessments were notable for blood eosinophil count of 120 cells/µL, a forced expiratory volume in 1 second (FEV1)/forced vital capacity (FVC) ratio of 0.63, and pre-bronchodilator FEV1 of 2.2 L (70% predicted).
Clara is a 71-year-old patient with COPD, who is uncontrolled, on maximum inhaled therapy, and she has elevated eosinophils. Her current symptoms include cough, chest tightness, sleep disruption, fatigue, and anxiety about having exacerbations. She thinks her symptoms are due to her age. Her BMI is 29 kg/m2, and her laboratory assessments are notable for blood eosinophil count of 480 cells/µL, FEV1/FVC ratio 0.67, and pre-bronchodilator FEV1 1.8 L (65% predicted).
Rabe then discussed how inflammatory changes, structural changes, and dynamic changes contribute to the impairment seen in patients with COPD, which is increasingly understood to be driven by chronic inflammation and structural changes. Chronic inflammation may be triggered by tobacco smoke, toxic particles or gases, viruses or bacteria, or oxidative stress.1-3 This inflammation drives structural changes in the airways (Figure 1). Compared with healthy airways, the airways of patients with COPD are characterised by barrier disruption, goblet cell hyperplasia, airway remodelling, inflammation, and excess mucus. Chronic inflammation also results in parenchymal destruction where alveolar membranes break down, and air trapping occurs. The combination of structural changes in the airways and parenchymal destruction results in airflow limitation.1-5
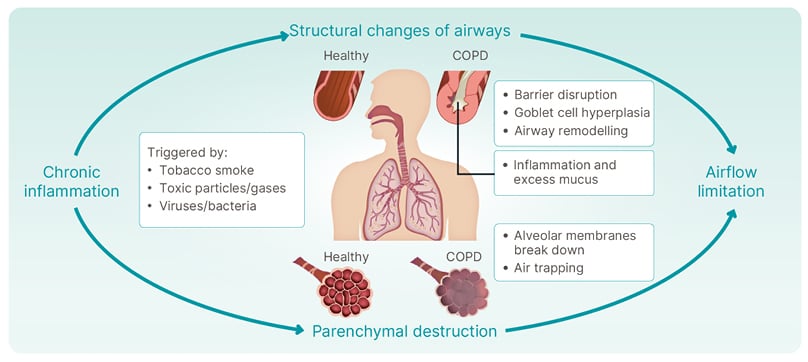
Figure 1: Chronic obstructive pulmonary disease is driven by chronic inflammation and structural changes.
COPD: chronic obstructive pulmonary disease.
Pioneering research conducted by Hogg et al. showed that small airways (<2 mm in internal diameter) are the primary site for airflow limitation in COPD.6 Compared with healthy individuals, the small airways of patients with COPD feature mucus plugging, increased synthesis of mucin genes, and goblet cell hyperplasia and mucus production.6-10 But what does the clinical presentation look like? In a hypothetical patient like Leonard, this may present as a cough with sputum and shortness of breath. In a hypothetical patient like Clara, this might present as chest tightness. Notably, current smoking is associated with goblet cell hyperplasia in patients with COPD.11
Furthermore, small airways disease has implications for dynamic hyperinflation, commonly observed in patients with COPD. In dynamic hyperinflation, small airways disease causes expiratory airflow limitation, where the inhaled volume of air exceeds the exhaled volume of air, with a progressive increase in functional residual capacity and a decrease in inspiratory capacity.12 Clinically, this may present as shortness of breath, impaired and abnormal gas exchange, respiratory and peripheral muscle dysfunction, as well as hypercapnia, respiratory acidosis, and impaired cardiopulmonary interaction due to hypoxemia and retention of carbon dioxide.12,13 In a hypothetical patient like Leonard, his chief complaints may be a cough and shortness of breath, while a hypothetical patient like Clara may exhibit symptoms like a cough and chest tightness. More importantly, there are downstream consequences like exercise avoidance and reduced health-related quality of life (HRQoL) for patients with COPD.12
Exercise avoidance and reduced HRQoL are particularly relevant in patients with COPD and persistent symptoms. Respiratory symptoms, such as breathlessness, cough, and excess sputum, are associated with a reduction in physical activity. Symptoms are also associated with poor HRQoL, anxiety, depressed mood, and sleep disturbance.2,14-16 In addition, a reduction in physical activity has implications for HRQoL, mood, and sleep, and vice versa.14 In a hypothetical patient like Leonard, persistent symptoms of COPD may manifest as shortness of breath when climbing or using stairs, while Clara may complain of sleep disturbance and fatigue, both affecting her quality of life.
The concept of COPD as a disease limited to the lungs, airways, and lung parenchyma has evolved. It is well known that systemic inflammation is present in COPD, including both pulmonary and extrapulmonary systems. Pulmonary co-existing conditions present in patients with COPD are, among others, asthma, bronchiectasis, and pulmonary hypertension. Importantly, patients with COPD have a significantly increased risk of lung cancer.2,17,18 Extrapulmonary comorbidities include, among others, cardiovascular disease, osteoporosis, sleep disorders, anxiety, depression, metabolic syndrome, renal failure, and gastrointestinal disease.2,17-19
Importantly, worsening disease leads to a vicious cycle of COPD. Starting with initiating factors, this cycle descends into chronic inflammation and structural changes, with subsequent symptoms and COPD exacerbations at the centre of the cycle.1-3,20 Exacerbations lead to a further decline in lung function and prolonged recovery, along with elevated risk of mortality, further perpetuating the cycle of progressive disease.2,21-26
In summary, Rabe provided a comprehensive overview of COPD, emphasising the heterogeneity in its clinical presentation, which stems from the complex interplay of underlying disease mechanisms, risk factors, and environmental exposures. He highlighted how chronic inflammation drives structural changes in the airways, which manifests as airflow limitation leading to shortness of breath, a main COPD symptom, and increased exacerbation risk. Because of the symptoms and exacerbations present in COPD, patients experience a significant impact on their activities of daily living, limitations in physical activity, worsened HRQoL, and multimorbidities, all of which worsen as their COPD worsens. From a clinical perspective, the variability in COPD presentations underscores the need for tailored management strategies that account for the diverse facets of this complex disease.
Heterogeneity of Inflammatory Pathways in Chronic Obstructive Pulmonary Disease
Stephanie Christenson
Increased understanding of COPD has highlighted the existence of diverse inflammatory pathways in the lungs, with inflammation that can be differentiated by distinct inflammatory cells and cytokines. Type 1/Type 3 inflammation is characterised by elevated sputum neutrophils, elevated Type 1 cytotoxic T cells, T helper (Th)1 cells, and Th17 cells; elevated group 3 innate lymphoid cells; elevated cytokines, such as IL-1β, TNF-α; and poor response to corticosteroids.27,28 Type 1/Type 3 inflammation is observed in the majority of patients with COPD.29 COPD with Type 2 inflammation is characterised by elevated sputum eosinophils; elevated Th2 cells; elevated group 2 innate lymphoid cells; elevated Type 2 cytokines, such as IL-4, IL-13, and IL-5; and responsiveness to corticosteroids.27,30-32 Type 2 inflammation is believed to comprise an estimated 30–40% of patients with COPD, based on sputum eosinophils ≥3% and blood eosinophils ≥2%.33,34 Notably, there is potential overlap in Type 2 and Type 1/Type 3 inflammatory mechanisms driving COPD.
Figure 2 illustrates the diverse inflammatory pathways associated with COPD, recognising that COPD is a complex disease, and not all pathophysiological pathways depicted may be specific to COPD. Viruses, bacteria, smoke, and/or pollutants can cause damage to an otherwise healthy airway epithelium via oxidative stress. The exposure triggers an inflammatory response, releasing alarmins (i.e., IL-25, IL-33, and thymic stromal lymphopoietin) from the airway epithelium and blood vessel endothelium. This process is common to both COPD with Type 2 and COPD with Type 1/Type 3 inflammation, with downstream inflammatory signalling. The chronicity leads to airway remodelling and fibrosis of the small airways, wall thickening, and mucus hyperproduction, which provides, at least partially, the clinical scenario of COPD.3-5,29,35-44
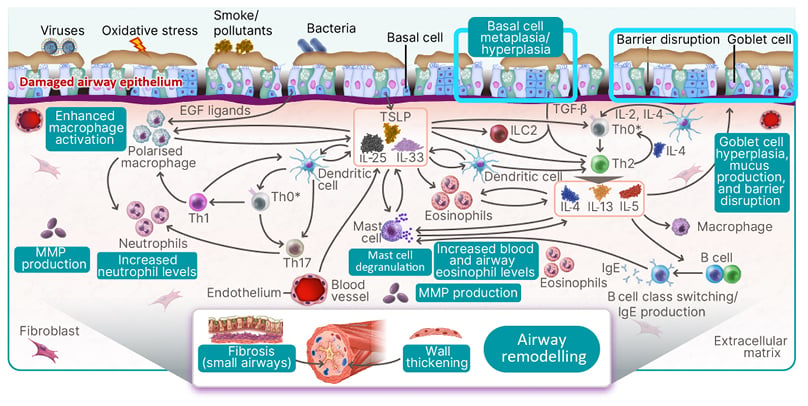
Figure 2: The inflammatory pathways associated with chronic obstructive pulmonary disease are diverse.
*T cell differentiation takes place in lymph nodes and is included in the schematic for the purposes of illustration only.
EGF: epidermal growth factor; ILC2: group 2 innate lymphoid cell; MMP: matrix metalloproteinase; Th: T helper cell; TSLP: thymic stromal lymphopoietin.
Specific to Type 1/Type 3 inflammation, the release of alarmins triggers several changes. Activated dendritic cells drive Th0 differentiation into Th1 and Th17 cells. Furthermore, epidermal growth factor ligands, alarmins, and IL-17 from Th1 cells enhance macrophage activation from the non-polarised to the polarised state. These polarised macrophages release IL-1β and TNF-α and, along with IL-17 released from Th17 cells, stimulate matrix metalloproteinase (MMP) production, and increase neutrophil levels.3,18,30,36-47
This is in comparison to Type 2 inflammation associated with COPD. The initial release of alarmins triggers Th0 differentiation into Th2 cells, mediated by dendritic cells, group 2 innate lymphoid cells, and TGF-β. Subsequently, Th2 cells release key cytokines (IL-4, IL-13, and IL-5), leading to a spectrum of outcomes.3,5,29,35,36,38,39,41,43 Pathologically, these cytokines induce goblet cell hyperplasia, heightened mucus production, disruptions in barrier integrity, and basal cell metaplasia/hyperplasia. Clinically, these pathological changes culminate in the hallmark symptoms of COPD, notably sputum production, persistent cough, and increased breathlessness. Furthermore, the key cytokines IL-4, IL-13, and IL-5 trigger elevated levels of eosinophils in both blood and airways. Moreover, they stimulate B cell class switching, triggering increased production of IgE, which, in turn, directly and indirectly prompts mast cell degranulation, and the release of additional inflammatory mediators. Additionally, MMP production and extracellular matrix degradation, though primarily observed in mouse models, represent another facet of the pathophysiological picture. Together, these pathophysiological changes have been associated with decreased FEV1 and broad remodelling, and can contribute to the distinct clinical features of COPD, such as worsening of respiratory symptoms.3,5,29,35-39,41,43
Notably, the potential role of Type 2 cytokines in COPD with Type 2 inflammation continues to be elucidated, and IL-4, IL-13, and IL-5 have distinct and overlapping effects on cell types involved in the pathophysiology of, and clinical outcomes observed in, COPD (Figure 3). Specifically, IL-4 is the key cytokine that induces the differentiation of naïve T cells into Th2 cells. Additionally, IL-4 stimulates basophil and mast cell degranulation.45 IL-13 plays a distinct role in goblet cell hyperplasia and mucus production.7,46,47 IL-5 has a distinct role in eosinophil activation in the bone marrow.48 IL-4 and IL-13 have overlapping effects on B cell class switching and IgE production, barrier disruption, airway remodelling (e.g., fibrosis, wall thickening, and MMP production), and alveolar destruction and airway enlargement. All three key cytokines play a role in stimulating eosinophil trafficking to tissues.39,45,49-51 Clinically, this range of effects results in the characteristic airflow limitation seen in patients with COPD. With chronic inflammation, patients experience persistent symptoms (shortness of breath, chronic cough, sputum production), progressive lung function decline, and COPD exacerbations.2
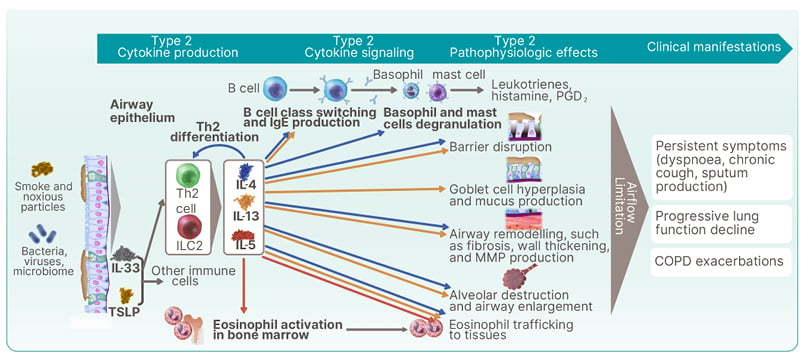
Figure 3: Potential roles of Type 2 cytokines in chronic obstructive pulmonary disease with Type 2 inflammation.
COPD: chronic obstructive pulmonary disease; ILC2: group 2 innate lymphoid cell; MMP: matrix metalloproteinase; PGD2: prostaglandin D2; Th2: T helper cell Type 2; TSLP: thymic stromal lymphopoietin.
In summary, Christenson highlighted the latest knowledge on the diverse inflammatory processes and pathways; the contribution of different pathways in causing pathologic changes; and the association between inflammation, pathologic changes, and clinical presentation in COPD. While there is some overlap in the inflammatory processes in Type 1/Type 3 inflammation and Type 2 inflammation, there are distinct differences that have consequences for disease presentation, clinical presentation, and potentially for management. In addition, the heterogeneity of inflammatory pathways in COPD highlights the need for continued investigation to better understand the disease, and facilitate a more targeted management approach for patients.
The Evolving Clinical Landscape in Chronic Obstructive Pulmonary Disease and Implications for Disease Management
Dave Singh
The clinical landscape in COPD continues to evolve rapidly, with implications for disease management. COPD classification for treatment continues to be guided by symptoms and exacerbations, as outlined in the 2023 Global Initiative for Chronic Obstructive Lung Disease (GOLD) ABE tool for initial assessment. Lung function confirmation is mandatory for the diagnosis of COPD (post-bronchodilator FEV1/FVC <0.7) and initial severity classification: GOLD Grade 1 defined as FEV1 ≥80% predicted; GOLD Grade 2 defined as 50–79% predicted; GOLD Grade 3 defined as 30–49% predicted; and GOLD Grade 4 defined as <30% predicted. Patients with COPD categorised as an ‘A’; are characterised by 0 or 1 moderate exacerbations (not leading to hospitalisation), and low symptoms defined by a modified Medical Research Council (mMRC) score of 0–1 and COPD Assessment Test (CAT) score <10. Patients with COPD categorised as a ‘B’ have the same exacerbation history as patients categorised as an ‘A’, but with higher scores on mMRC (≥2) and CAT (≥10). Patients with COPD categorised as ‘E’ experience ≥2 moderate exacerbations or ≥1 leading to hospitalisation, regardless of symptoms, based on mMRC or CAT score.2
The future is a more targeted approach, based, in addition to these features, on biological understanding and other phenotypic and endotypic characteristics, which may link to some of the heterogeneous inflammatory pathways underpinning COPD, as discussed by Christenson. Singh highlighted several possible mechanisms that are under investigation, and in various stages of clinical development. While it is not possible to delve into the details of the numerous clinical development programmes, it is notable that these programmes do encompass diverse COPD patient groups, which is particularly relevant for patients with COPD who experience exacerbations.
As highlighted in the GOLD 2023 follow-up algorithm, blood eosinophil counts can be used to identify patients for whom treatment should be escalated. For patients treated with LABA or LAMA, who exacerbate and have a blood eosinophil count of ≥300, their treatment regimen should consist of LABA plus LAMA plus inhaled corticosteroids (ICS). If these patients continue to exacerbate despite escalation, the available options are limited. For patients who experience an exacerbation and have a blood eosinophil count <300, their treatment regimen should be escalated to LABA plus LAMA. From there, if they continue to experience exacerbations with a blood eosinophil count <100, treatment options are limited. For those with a blood eosinophil count ≥100, the next step is triple therapy with LABA plus LAMA plus ICS, though ICS can be contraindicated in some patients.2 For a hypothetical clinical scenario like Leonard, he may have another treatment option following exacerbation on triple therapy. Subsequent treatment options are limited. In a hypothetical clinical scenario like Clara’s, uncontrolled on maximum inhaled therapy and with elevated eosinophils, there is an unmet need for additional treatment options after triple therapy.
Singh summarised by noting that COPD is a mixed disease with mixed pathology. For some patients, Type 2 inflammation might be the dominant feature, while for others it may not be. Rabe, Christenson, and Singh further discussed the current excitement around COPD research, the complexity of the disease, and the movement of the field towards a better understanding of patient phenotypes and endotypes. With the treatment landscape evolving, phenotyping patients will be crucial to optimising patient care.
Rabe concluded the symposium by thanking his co-panellists and the audience.
MAT-GLB-2304728 v1
10/2023