Meeting Summary
Understanding the role of the epithelial cytokines thymic stromal lymphopoietin (TSLP), IL-25, and IL-33 in inflammatory airway diseases such as severe asthma (SA), chronic rhinosinusitis with nasal polyps (CRSwNP), and chronic obstructive pulmonary disease (COPD) could lead to new insights into the pathophysiology of airway diseases. The airway epithelium plays a critical role in these diseases, with TSLP, IL-25, and IL-33 released following exposure to disease triggers, such as allergens, pollutants, and pathogens, which can exacerbate these conditions. These cytokines can also be released from both immune and non-immune cells and can act on a broad array of target cells. In asthma, the release of TSLP, IL-25, and IL-33 following exposure to a trigger has been linked to airway inflammation and hyperresponsiveness. Epithelial disruption in CRSwNP can drive inflammation, impair mucociliary clearance, and increase permeability and mucus production. Epithelial cytokine elevation is especially prominent in eosinophilic CRSwNP, where it is positively correlated with symptoms and underlying pathophysiology. Conversely, causative mechanisms in COPD are poorly understood, and management is sometimes inadequate. However, similar to asthma, IL-33 and TSLP are increased in patients with COPD, indicating that they may also have a role in the pathophysiology of this disease. Significant challenges remain regarding understanding of SA, CRSwNP, and COPD, especially in patients where symptoms are uncontrolled. Therefore, elucidating the role of TSLP, IL-25, and IL-33 in these conditions may have the potential to offer new insight into these conditions.
Introduction
Understanding of the airway epithelium initially focused on its role as a protective barrier and environmental sensor. However, it is now known that numerous, and in some individuals, multiple, environmental triggers can initiate epithelial-driven innate and adaptive immune responses.1 These can lead to the inflammatory changes seen in acute and chronic obstructive airway diseases, structural changes, and airway remodeling (Figure 1).2-5 As such, the airway epithelium is becoming a focus for elucidating the pathophysiology of inflammatory conditions such as SA, CRSwNP, and COPD.
While descriptions of upper and lower airway diseases have been noted for centuries, elucidation of their pathogenesis has only occurred in the last 150 years, with more advanced descriptions of associated inflammatory signaling stemming from the mid-1980s.6-8 One such development has been additional understanding of the role of three airway epithelium-produced proinflammatory cytokines with roles in SA, CRSwNP, and COPD: TSLP, IL-25, and IL-33.9 While these diseases have their own manifestations, findings regarding concurrence of global prevalence, similar cell and tissue type representation, and shared phenotypes support a unified airway model regarding their pathogenesis,10 which may involve these epithelial cytokines.11
Human TSLP was first identified in 2001,12 with its role in activation of a key cell type involved in airway diseases, Type 2 innate lymphoid cells (ILC2), not described until 2016.13 IL-25 was also first described in 2001,14 with description of IL-33 in human endothelial cells dating to 2003.15 These epithelial cytokines can activate inflammatory responses in both distinct and overlapping ways, and may have implications in the underlying inflammatory processes in SA, CRSwNP, and COPD (Figure 1).16
While the airway epithelium is the primary source of TSLP, IL-25, and IL-33, they can also be produced by a range of both immune cells (Figure 1) and non-immune cells, such as stromal cells, fibroblasts, and smooth muscle cells. These epithelial cytokines can also target a wide array of cells, leading to complex, bi-directional cell–cell interactions.16-20 Following activation of inflammatory cells by TSLP, IL-25, and IL-33, proinflammatory cytokines expressed by the affected cells include IL-4, IL-5, IL-9, IL-13, and granulocyte macrophage colony stimulating factor.2,16-20 These mediators can contribute to upper and lower airway disease development and pathology, including inflammation, airway hyperresponsiveness, mucus production, and potentially, airway remodeling.20-22
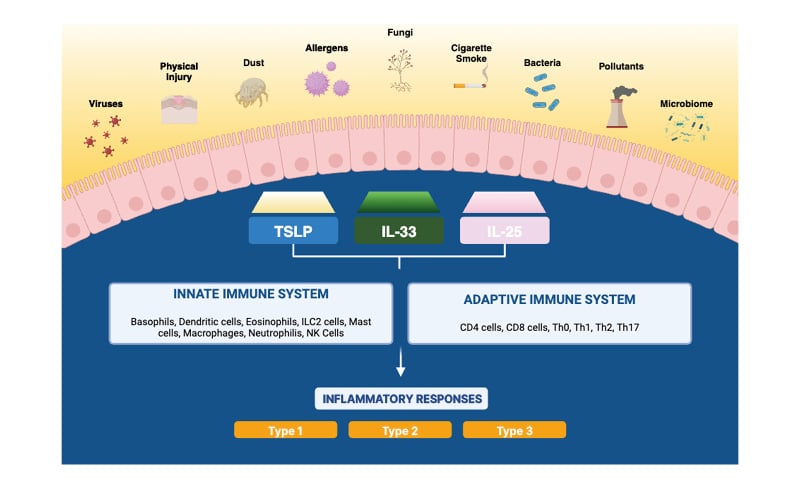
Figure 1: The role of TSLP, IL-25, and IL-33 in airway diseases.
ILC2: Type 2 innate lymphoid cell; NK: natural killer; Th: T-helper; TSLP: thymic stromal lymphopoietin.
Severe Asthma
Around 262 million people worldwide are affected by asthma, a condition involving excessive bronchoconstriction due to airway inflammation, airway hyperresponsiveness (which can occur regardless of the level of airway inflammation), and, in some patients, reduced lung function.23 Asthma is classed as severe in around 5−12% of people with the condition.24 One Canada-based study found that, compared to non-severe asthma, SA accounts for at least half of all asthma-related excess healthcare costs, especially when it is comorbid with other medical conditions.25
Patients with severe asthma can have multiple asthma triggers (Figure 1). For example, in a study by Chipps and colleagues,1 patients had on average eight different triggers. This study also found that as trigger number increases, disease control can lower, asthma exacerbations and hospitalizations can increase, work productivity can decrease, and health-related quality of life can worsen.1 Most patients with SA show activation of multiple inflammatory pathways and their respective biomarkers. For example, a study of patients from the International Severe Asthma Registry (ISAR) found that, of 1,175 adults with SA, nearly all (88%) had between 1-3 biomarkers for asthma, as evidenced by allergen-specific serum IgE positivity ≥75 kU/L, fractional exhaled nitric oxide levels ≥25 parts per billion, or blood eosinophils ≥300 cells/mL).26
Similarly, a post hoc analysis of multiple Phase IIb/III trials found that, of 740 participants with SA with blood eosinophils ≥150 cells/mL, nearly 80% had allergic asthma, and nearly 50% had fractional exhaled nitric oxide levels ≥25 parts per billion.27
Release from the airway epithelium of TSLP, IL-25, and IL-33, and subsequent activation of multiple inflammatory pathways2,16,21 is linked to several hallmarks of SA, including airway hyperresponsiveness28 and remodeling,29 decreased lung function,30 and increased asthma severity,30,31 exacerbation risk,32 and mucus production.17,33
The epithelial cytokines IL-25, IL-33, and TSLP may play a role in initiation and persistence of airway inflammation, leading to manifestations of asthma. For example, allergen challenge can significantly increase the number of bronchial epithelium and submucosa cells expressing these epithelial cytokines,28 and airway concentrations of TSLP and IL-33 are significantly increased in patients with asthma, particularly SA, compared with healthy controls.30 Airway inflammation can contribute to lower lung function, and a significant inverse correlation has been shown between forced expiratory volume in 1 second and both TSLP and IL-33 levels in bronchoalveolar lavage fluid, and the number of bronchial epithelium and submucosa cells reactive for these cytokines.28,30
TSLP and IL-33 levels are also significantly increased in patients with asthma who use inhaled corticosteroids (with or without oral corticosteroids) compared with patients who do not use these treatments, indicating that levels of these epithelial cytokines increase as the need for medication increases.30,31 Additionally, higher serum TSLP levels (≥25 pg/mL) are associated with higher exacerbation risk in SA, suggesting that TSLP appears to be involved in the pathogenesis of asthma exacerbations.32
Indications that these cytokines have a role in downstream pathways associated with asthma induction and severity come from several investigations. For example, upregulation of OX40 ligand on dendritic cells by epithelium-derived TSLP can induce an interaction with OX40 on T cells, polarizing them to a Type 2 phenotype and leading to the release of IL-4, IL-5, and IL-13.2,16,22
Levels of these inflammatory cytokines correlate significantly with lung function in asthma.30 They are involved in SA-linked pathologies including nitric oxide induction, IgE production, goblet cell hyperplasia (goblet cell hyperplasia leads to mucus hypersecretion and may be involved in mucus plug formation), and smooth muscle reactivity, linked to bronchial hyperresponsiveness 2,21 Additionally, IL-5 is the primary growth factor for eosinophils, and upregulation by IL-5 promotes eosinophilic inflammation.2,22
Chronic Rhinosinusitis with Nasal Polyps
As a gateway to the respiratory system, nasal airway epithelium is often the first point of contact for microorganisms, pollutants, and allergens.3 Although the immune response launched by airway epithelial cells is important for defense, it can also drive inflammatory diseases. For example, in allergic rhinitis, allergen exposure drives IgE-mediated mucosal inflammation that can become chronic.3 Chronic rhinosinusitis, generally categorized into occurrence with or without nasal polyps, is diagnosed in patients with symptoms and evidence of sinus inflammation on nasal exam or imaging that has persisted for at least 12 weeks.34
CRSwNP, which typically occurs between ages 40−60 years,35 is estimated to globally affect up to 4.4% of the population, with a slightly higher prevalence in males.35,36 Cardinal signs and symptoms of CRSwNP include nasal congestion, loss of sense of smell, and impaired sleep quality, in addition to facial pain and pressure and nasal discharge. Loss of the sense of smell is postulated to be the result of blockage of access, along with inflammation driven damage, to the olfactory tissue.34,35
Based on a USA-based study, compared with patients without chronic rhinosinusitis, patients with CRSwNP are estimated to visit an emergency department 1.5 times as often, a healthcare professional 2.0 times as often, and have incremental healthcare costs 2.5 times greater.37 Troublesome symptoms of CRSwNP, such as nasal congestion and loss of the sense of smell,38,39 can have not only physical but also mental and social impacts on health-related quality of life.39 Despite such findings, patients with CRSwNP express frustration due to perceived underestimation of the impact of the disease.38
Around two-thirds of patients with CRSwNP also have asthma, with this comorbidity leading to a greater burden of disease, including more difficulty in gaining asthma control and a higher likelihood of experiencing polyp recurrence and steroid dependence.11 The idea that CRSwNP and asthma share common underlying inflammatory pathways is supported by studies showing that allergen challenges in either upper or lower airway alone can lead to subsequent presence of inflammatory mediators in both locations. This may be linked to expression in both diseases of epithelium cytokines,40 supporting the unified airway model.10
As in SA, in CRSwNP, the epithelium is not just a passive barrier, with damage only caused by immune system cells, but is an intricate part of the pathophysiology of the disease. Damage to the sinus epithelium can trigger cell proliferation and transformation of epithelial cells to a mesenchymal form, leading to inflammatory processes and remodeling.41 In addition, upregulation of mast cell and basophil degranulation results in vasodilation, with leakage of plasma containing fibrin and edema. This is followed by accumulation of cross-linked fibrin, which forms the bulk of the nasal polyp, forming a dense mesh capable of trapping plasma proteins and water, leading to edema and remodeling.42
Three inflammatory endotypes of CRSwNP have been described, which can be mixed in many patients, or not present in a minority.43-45 Type 1 is characterized by the release of interferon-γ from ILC1, Th1 cells, cytotoxic T cells, and natural killer cells.44 Dominant clinical features include headache and facial pain.45 Cytokines released in Type 3 CRSwNP include IL-17 and IL-22 from ILC3s, Th17 cells, and neutrophils.44 Key features of this endotype include eye itching and purulent rhinorrhea.43-45
However, most patients with CRSwNP are Type 2, characterized by anosmia, asthma comorbidity, and high clinical burden.43-45 In this endotype, upregulation of TSLP, IL-25, and IL-3342 can lead to the release of IL-4, IL-5, and IL-13 from eosinophils, and Th2 cells, basophils, and ILC2s.42,44 Higher levels of these epithelial cytokines or markers for them have been found in nasal polyp tissue of patients with CRSwNP compared with uncinate process or turbinate tissue of controls.46-48 Studies also point to a role of some of these epithelial cytokines in conversion of ILC2 cells to a corticosteroid-resistant subtype.49 This is borne out by findings that TSLP levels are significantly increased in nasal epithelial cells in patients who have corticosteroid-resistant nasal polyps.50
The epithelial cytokines are particularly increased in eosinophilic CRSwNP (eCRSwNP) compared with controls and with patients with non-eCRSwNP.51 There are also correlations between disease severity in patients with eCRSwNP and expression of TSLP, the TSLP receptor, and the membrane-bound IL-33 receptor ST2L, based on patient ratings of symptoms and CT scan.52 Similar to SA, these findings suggest an intimate role of the epithelial cytokines in CRSwNP pathology.48,52
Chronic Obstructive Pulmonary Disease
Globally, an estimated 392 million people aged 30−79 years live with COPD,53 with numbers,54 along with healthcare-related costs,55 predicted to rise over the coming decades. The number of adults living in the USA with COPD is currently around 14.2 million, with little recent change in prevalence over the time period 2011 to 2021.56 In 2021, COPD was the sixth leading cause of death in the USA, with fatality numbers at nearly 140,000.57 Management options for COPD may not sufficiently target inflammatory pathways that contribute to this condition.4 This is evidenced by a number of findings, including that of a study that showed that half of hospital-treated patients with COPD exacerbations experienced treatment failure after 3 months.58
COPD is a complex, heterogeneous, and progressive disease,59 with myriad symptoms including airflow obstruction, mucus production, ciliary dysfunction, fibrosis, increased airway wall thickness, chronic bronchitis, fibrosis, and emphysema. Similar to SA and CRSwNP, COPD exacerbations can be brought about by multiple triggers, including infections, pollutants, cigarette smoke, and oxidative stress, leading to innate and adaptive immune pathway activation, inflammatory cell recruitment, cytokine/chemokine production, and elastase and matrix metalloproteinase activation.4,60
The mechanisms behind COPD development have not been fully elucidated; however, epithelial cytokines, TSLP and IL-33 have been suggested to play a role in the underlying inflammatory processes associated with the disease.61 For example, TSLP is significantly increased in bronchoalveolar lavage fluid,62 epithelial tissue,63 and airway smooth muscle63 in patients with COPD compared with both smoking and non-smoking controls.62-64 There are also correlations of TSLP with significantly increased platelet counts and higher platelet aggregation rates.65
Following bronchial epithelium injury, epithelial cytokine secretion, including TSLP, is increased, activating multiple cell types implicated in COPD pathophysiology, including T cells, ILC2s, and eosinophils.61 TSLP is also implicated in changes in non-immune system cells associated with COPD. For example, studies have shown higher expression of the TSLP receptor on fibrocytes from people with eosinophilic COPD compared with non-eosinophilic COPD,66 and, in an in vitro study of human lung fibroblasts, TSLP upregulated collagen Type 1 and matrix metalloproteinase 1.67 IL-33 can be released from the airway epithelium in response to COPD-related triggers, such as cigarette smoke, microbes, and pollutants,2 and compared with healthy controls, levels of IL-33 in exhaled breath condensate are significantly higher in patients with non-atopic COPD.68
Further, higher levels of IL-33 in plasma are significantly associated with an increased risk of COPD exacerbations, and intensity of IL-33 immunostaining in lung parenchyma is inversely correlated with the forced expiratory volume in 1 second in such patients.69
Of note, cigarette smoking, a major risk factor for the development of COPD,59 can also upregulate the expression of IL-33, with expression of this epithelial cytokine significantly correlated with smoking history.69 However, levels of IL-33 in serum and sputum are significantly higher in patients with COPD compared with healthy non-smokers without COPD.68 These findings, along with those of TSLP discussed above, suggest that, while smoking may increase epithelial cytokines independently of COPD, the levels in patients with this condition are above that of those caused only by cigarette smoke inhalation.
Conclusion
For SA, CRSwNP, and COPD, there are still a number of outstanding factors to explore when assessing disease pathogenesis and control. Airway epithelium plays a critical role in these conditions, with TSLP, IL-25, and IL-33 released following immune cell activation or epithelial damage. These cytokines can also be released by, and act on, a wide array of both immune and non-immune cells.2 While TSLP, IL-25, and IL-33 have been linked to the pathogenesis of SA, CRSwNP, and COPD,15,16,20 further understanding of their role in these diseases is needed through both mechanistic and pre-clinical studies.