Abstract
Although thymomas and thymic carcinomas only represent 0.2–1.5% of all malignancies, they are the most common tumour found in the anterior mediastinum. Recently, the World Health Organization (WHO) classification of thymic epithelial tumours was revised and a new tumour, node, and metastasis (TNM) staging system is currently being developed. Nearly a third of patients with thymoma present with paraneoplastic syndromes, most commonly myasthenia gravis. Thymic carcinomas are rarely associated with paraneoplastic syndromes, with patients often presenting with local symptoms. Recommendations for the management of these tumours are primarily based on small prospective studies, meta-analyses, and expert guidelines. The development of novel therapies to treat thymomas and thymic carcinomas is an area of robust research.
INTRODUCTION
The differential diagnosis of a mediastinal mass is broad, including malignant and benign aetiologies, such as lymphomas, germ cell tumours, thymic tumours, thyroid goiter, infections such as tuberculosis, and granulomatous disorders such as sarcoidosis. Although rare, thymic epithelial tumours, thymomas, and thymic carcinomas are the most common primary tumours in the anterior mediastinum.1 While there exists some morphologic overlap between thymomas and thymic carcinomas, the two diseases differ in their clinical presentation, natural history, and recommended treatment strategies. Thymomas are chemo-sensitive and often follow an indolent course whereas thymic carcinomas are associated with a suboptimal response to conventional chemotherapy, high risk of recurrence, and inferior survival. Thymic epithelial tumours are reviewed in this paper, including a brief discussion of novel systemic therapeutic options.
EPIDEMIOLOGY
Thymomas account for about 20% of mediastinal neoplasms and about 50% of primary anterior mediastinum neoplasms.2 Nevertheless, these are uncommon tumours, with an estimated overall incidence in the USA of 0.13 per 100,000 person-years.3 Incidences are similar in males and females, increasing from 40 years of age and peaking at 70 years.3 Thymomas are more common and present at an earlier age among African Americans and Asians/Pacific Islanders compared to Caucasians. Racial variations in incidences and age at diagnosis suggest a genetic contribution, though the mechanism is unclear.2,3
Thymic carcinomas are very rare, accounting for <20% of thymic neoplasms and <100 cases in the USA per year.4,5 They occur predominantly in Caucasians.5 Thymic carcinomas occur over a wide age range, including adolescence, with their peak prevalence in the sixth decade of life.5
Given the rarity of thymomas and thymic carcinomas, the knowledge of their risk factors is limited. An association between tobacco or alcohol use and an increased risk of thymic carcinoma has not been established. Epidemiologic data do not suggest a link to occupational or environmental factors.
PATHOLOGY
There are many different classification schemes for thymic epithelial tumours. The World Health Organization (WHO) Classification6 was published in 1999 to standardise the classification of thymic tumours (Table 1). Suster and Moran7 also proposed a classification system in 1999 that addressed some of the perceived flaws of the WHO classification. The Suster and Moran Classification grouped thymic tumours into well-differentiated tumours (thymomas), moderately differentiated tumours (atypical thymomas), and poorly differentiated tumours (thymic carcinomas). This classification was felt to better reflect the prognosis of thymic tumours.
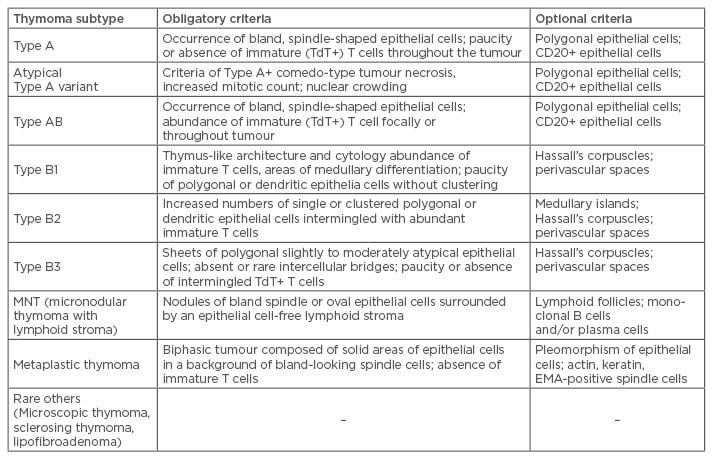
Table 1: Refined diagnostic criteria of thymomas in the 2015 World Health Organization (WHO) classification.
Adapted from Suster S, Moran CA.7
Despite its shortcomings, the WHO histologic classification is currently the most widely used classification system of thymic epithelial tumours and was most recently revised in 2015.6 The latest version focusses on histologic and immunohistochemical diagnostic criteria for thymomas and the distinction between thymomas and thymic carcinomas (Table 1).8 Immunohistochemical features are included in the diagnostic criteria of otherwise difficult-to-classify thymomas and thymic carcinomas.7 The WHO classification system emphasises that major thymoma subtypes, regardless of stage, should no longer be considered benign, as all potentially behave aggressively.8
There are many different subtypes of thymic carcinomas, including squamous cell, basaloid, mucoepidermoid, sarcomatoid, adenocarcinoma, nuclear protein in testis carcinoma, and undifferentiated carcinoma. These cancers need to be differentiated from metastatic lesions, and while immunohistochemical features can be helpful, clinical staging is very important.6
CLINICAL PRESENTATION
Thymic epithelial tumours present as an incidental finding on imaging studies because of local symptoms related to the bulk of the mediastinal mass, or because of a concurrently diagnosed paraneoplastic syndrome. Approximately one-third of patients diagnosed with thymoma are asymptomatic at time of presentation. With the growing use of computed tomography (CT) scans for diverse conditions, as well as screening for lung cancer, the number of asymptomatic patients diagnosed with thymomas is increasing.9 Nearly 40% of patients present with symptoms related to the mass effect of the tumour, most commonly cough, chest pain, and dyspnoea. Cases complicated by superior vena cava syndrome are also occasionally reported.
Paraneoplastic syndromes, most frequently myasthenia gravis, are frequently associated with thymomas.1 A rich infiltration of abnormally conditioned T cells is thought to be responsible for the strong association between paraneoplastic syndromes and thymomas.3 Paraneoplastic syndromes may occur either before or after the diagnosis of thymoma, or even following treatment. A population-based epidemiologic study using data from the Nationwide Swedish Cancer Registry revealed that thymoma patients were more likely to have an autoimmune disease at some point in their lifetime compared with controls (32.7% versus 2.4%; p<0.001). Among autoimmune diseases, myasthenia gravis (24.5%), systemic lupus erythematosus (2.4%), pure red cell aplasia (1.2%), sarcoidosis (0.9%), and rheumatoid arthritis (0.7%) were frequently observed.10
Although both thymomas and thymic carcinomas originate in the epithelial tissue of the thymus, their clinical presentations are quite different.11 Thymic carcinomas tend to behave more aggressively than thymomas, and approximately 80% of patients present with evidence of invasion of contiguous mediastinal structures. This local invasion often results in cough, chest pain, phrenic nerve palsy, or superior vena cava syndrome.4 Furthermore, paraneoplastic syndromes such as myasthenia gravis are very rarely associated with thymic carcinoma.5,12 If myasthenia gravis is present, the diagnosis of thymic carcinoma should be reassessed to rule out thymoma.13
DIAGNOSIS
The initial evaluation of a patient with a thymic tumour should include radiographic imaging with a contrast-enhanced CT scan of the chest. CT scans play a role in characterisation of the tumour, staging, and follow-up. Magnetic resonance imaging (MRI) scans are most helpful in cases of suspected infiltration of the heart and great vessels or to differentiate a thymic malignancy from a thymic cyst.14-16 Fluorodeoxyglucose positron emission tomography (FDG-PET) scans show significantly higher uptake of fluorodeoxyglucose among thymic carcinomas compared to thymomas.17 However, although using FDG-PET can be helpful in select cases, its use as a routine test has yet to be determined, and further research is awaited.18
Although the definitive diagnosis of a thymoma or thymic carcinoma requires a histological diagnosis, the need for biopsy prior to treatment depends on tumour resectability. Patients with tumours amenable to complete resection should undergo surgery, which will establish a definitive diagnosis as well as provide a therapeutic benefit. If the tumour cannot be completely resected, or if lymphoma is considered a likely diagnosis, then a histologic diagnosis with a core needle biopsy or an open biopsy should be performed prior to any systemic therapy or radiation.1 As thymic carcinomas are predominantly squamous cell or undifferentiated carcinomas, they should be differentiated from primary carcinoma of the lung, which can metastasise to the thymus and have a similar histologic appearance.
STAGING
The modified Masaoka–Koga staging system is widely used for both thymomas and thymic carcinomas (Table 2). In large studies, this system has been shown to correlate well with overall survival.5 A tumour, node, metastasis (TNM)-based staging system was proposed through collaboration of the International Association for the Study of Lung Cancer (IASLC) and the International Thymic Malignancy Interest Group (ITMIG) in 2014. Their intention was to develop an official and consistent staging classification system. A worldwide retrospective database was created by ITMIG, which represented the collaborative effort of 105 institutions and included 10,808 patients.19 However, correlative clinical data based on this system is still lacking and awaited. For this review, future mention of stage will refer to the Masaoka–Koga classification.
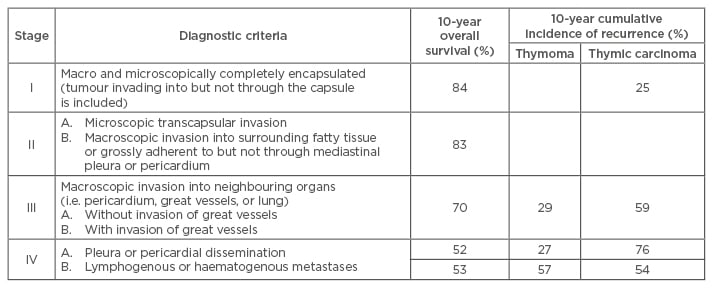
Table 2: Modified Masaoka–Koga Clinical Staging of thymoma with overall survival and recurrence-free survival.
Adapted from Falkson et al.11 and Girard et al.20
MANAGEMENT
The treatment strategies for treatment adapted from National Comprehensive Cancer Network (NCCN) and European Society for Medical Oncology (ESMO) guidelines are outlined in Figure 1.20 Given the absence of randomised data to guide management, recommendations are based primarily on retrospective data and expert opinion.
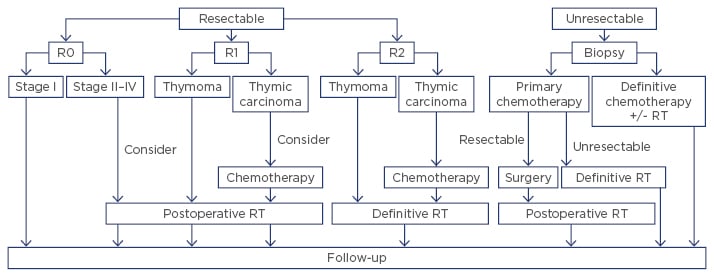
Figure 1: Treatment strategy of thymic tumours.
RT: radiation therapy.
Adapted from Ettinger et al.13 and Girard et al.20
Surgery
Surgical intervention is the mainstay of treatment; thus, assessment of respectability is the first step in the treatment of thymic epithelial tumours. Total thymectomy with complete surgical excision of the tumour is crucial to achieve cure in appropriate candidates. Although a recent meta-analysis showed similar effectiveness of minimally invasive surgery compared to open thymectomy in selected patients,21 a minimally invasive approach is not routinely recommended due to the paucity of robust clinical data confirming a benefit in long-term prognosis. Therefore, both NCCN13 and ESMO guidelines20 recommend consideration of minimally invasive surgery for clinical Stage I–II in limited settings, including availability of appropriately trained thoracic surgeons. Partial thymectomy is also an option for Stage I tumours in patients without myasthenia gravis.22 Historically, lymphadenectomy was not routinely performed with thymectomy; however, lymphadenectomy should be considered for all thymic carcinomas given the higher risk for lymph node metastasis.23
Prior to any surgical procedure, all patients suspected to have thymomas, whether symptomatic or not, should undergo assessment of serum anti-acetylcholine receptor antibody levels. If patients have myasthenia gravis, a neurology evaluation should be obtained to avoid perioperative respiratory failure.24
Radiotherapy
Generally, thymic epithelial tumours are moderate to highly radiosensitive. As shown in Figure 1, radiotherapy plays a role as adjuvant or definitive therapy, either as a single agent or concurrent with chemotherapy. However, evidence supporting adjuvant radiation from multicentre, prospective trials is lacking; thus, recommendations are based on data from large cohorts and pooled analyses of retrospective studies.20 There is no known survival benefit associated with the delivery of postoperative radiotherapy in Stage I–III thymomas following R0–1 resection.12,25-30 Multiple studies have demonstrated a similar rate of recurrence after complete resection regardless of postoperative radiotherapy.26 One review of the National Cancer Institute (NCI)-sponsored SEER (Surveillance, Epidemiology, and End Results) programme database, which provides incidence and survival data from tumour registries across the USA, even suggested that adjuvant radiation for localised disease may have an adverse impact on survival.27 Recommendations for postoperative radiotherapy are reserved for patients with a higher risk of recurrence, such as those with a complete resection of a Stage III–IVA thymoma or an incompletely resected thymoma of any stage.5,26-28,31 Thymomas do not typically metastasise to regional lymph nodes; thus, extensive elective nodal radiotherapy is not recommended.32
Adjuvant radiation should also be recommended for all patients with completely or incompletely resected thymic carcinoma due to the high risk of recurrence.33,34 A recent meta-analysis of 973 patients with thymic carcinoma concluded that adjuvant radiation therapy offers both a progression free survival (hazard ratio: 0.54; 95% confidence interval: 0.41–0.71) and overall survival benefit (hazard ratio: 0.66; 95% confidence interval: 0.54–0.80).35
Postoperative radiotherapy should be initiated within 3 months after surgery via three-dimensional conformal techniques or intensity-modulated radiation therapy. Although the optimal dose is not defined, a total dose of 45–50 Gy for complete (R0) resection or 54 Gy for R1 resection is recommended. Following R2 resection or for definitive radiotherapy of unresectable or inoperable disease, a total dose of 60–70 Gy should be administered.
Chemotherapy
Generally, thymomas are considered chemo-sensitive, whereas chemotherapy has only modest activity against thymic carcinomas. Chemotherapy is administered both to reduce the risk of recurrence and to palliate symptoms. Adjuvant chemotherapy has not been shown to improve survival following R0–1 resection of thymomas.11,13,20,36 On the contrary, platinum-based postoperative chemotherapy can be considered for Stage II-IV thymic carcinomas given the high risk of recurrence, especially following incomplete resection.11,13,20
Induction chemotherapy to downstage the tumour prior to surgery is recommended for locally advanced, Stage III–IV thymic tumours for which upfront complete resection is deemed not achievable.12,13,20 Usually, 2–4 cycles of platinum-based chemotherapy (e.g. cisplatin, doxorubicin, and cyclophosphamide (CAP); cisplatin and etoposide; or carboplatin and paclitaxel) are administered followed by re-imaging for evaluation of surgical resectability.37 Cisplatin and etoposide concurrent with radiation can also be considered for unresectable localised tumours, either to downstage in anticipation of surgery or as definitive therapy.20
For patients with unresectable metastatic disease, palliative chemotherapy is administered to relieve tumour-related symptoms and prolong survival. Given the absence of randomised studies, broadly accepted optimal regimens and precise survival benefits are not available. Both NCCN and ESMO guidelines recommend the triplet therapy of CAP as the preferred first-line regimen for thymomas.13,20 A study of CAP in 29 patients with thymoma and 1 with thymic carcinoma revealed a 50% response rate and 37.7-month overall survival.38 For patients who cannot tolerate a cisplatin and anthracycline-based regimen, carboplatin and paclitaxel can be considered. A platinum-taxane regimen is also recommended as first-line treatment for patients with thymic carcinoma.13,20,39 A Phase II study of carboplatin and paclitaxel in 46 patients demonstrated response rates of 42.9% and 21.7% for thymoma and thymic carcinoma, respectively.40
NOVEL SYSTEMIC THERAPIES
The limited effectiveness of cytotoxic chemotherapy in advanced thymic carcinoma, and the lack of evidence-based treatment options for relapsed or refractory thymoma, has meant that the development of novel agents has been an active area of research. Somatostatin receptors are often expressed in thymic epithelial tumours, and these tumours are detectable by octreotide scan. Octreotide administered via subcutaneous injections demonstrated modest activity (30.3% objective response rate) in a small Phase II trial.41 Of note, none of the thymic carcinoma patients included in the trial demonstrated a response to octreotide.
Small molecule tyrosine kinase inhibitors have also been studied in previously treated thymic tumours. An analysis of thymic carcinomas demonstrated the activation of receptor tyrosine kinases, including platelet-derived growth factor receptor-beta and vascular endothelial growth factor receptor-3, both targets of sunitinib.42 A Phase II trial of sunitinib in relapsed and refractory thymic epithelial tumours demonstrated a 26% response rate among thymic carcinomas.43
More recently, a study of the anti-programmed cell death (PD)-1 checkpoint inhibitor, pembrolizumab, was reported at the 2017 American Society of Clinical Oncology (ASCO) Annual Meeting. Investigators described a 22.5% objective response rate among patients with refractory thymic carcinoma. A response was detected in 6 of 9 patients with high PD-L1 expression, though a correlation between tumour mutational burden and pembrolizumab activity was not noted.44 Immune-related adverse effects were observed at a higher than expected rate with 6 of the 40 patients experiencing Grade 3–4 immune-related adverse effects, including two noted to have myocarditis.
Beyond cytotoxic chemotherapy, a number of medications with different biological mechanisms have activity against refractory thymic epithelial tumours. Larger studies and the identification of predictive biomarkers are needed prior to moving these treatments into the armamentarium of routine clinical practice.
PROGNOSIS
As thymomas usually are indolent in nature, the extent of invasion and the completeness of resection have the greatest impact on prognosis.45 The prognostic value of tumour histology remains controversial.46,47 For resected Stage I and II thymomas, 10-year survival rate is excellent and >80% (Table 2).
Survival rates for thymic carcinomas, are also heavily influenced by stage and resectability. A large series of patients with thymic carcinoma in North America (N=121) showed 5-year overall survival of 100%, 81%, 51%, 24%, and 17% for Stage I, II, III, IVa, and IVb, respectively.5
FOLLOW-UP
There are no prospective data available to establish recommendations for post-treatment surveillance. Generally, prolonged follow-up indicated as late relapse is possible and a relapse may be potentially treated with curative-intent. NCCN guidelines recommend a chest CT every 6 months for 2 years, then annually for 5 years for thymic carcinomas, and for 10 years for thymomas.13,48 ESMO clinical practice guidelines recommend baseline chest CT 3–4 months after surgery, followed by an annual chest CT for completely resected Stage I/II thymomas for 5 years, and then every 2 years. For Stage III/IV thymomas, thymic carcinomas, or after a R1/R2 resection, the guidelines propose chest CT every 6 months for 2 years and then annually. The recommended duration for imaging follow-up is 10–15 years.20 Subsequent malignancies and/or late onset paraneoplastic syndrome may occur as well; however, there is no clear guidance of follow-up for these phenomena.13,20