Abstract
Lance-Adams syndrome (LAS) is a rare complication that occurs after successful cardiopulmonary resuscitation, in which myoclonus appears rapidly upon recovery from the severe hypoxic event. To date, <150 cases have been reported. The author reports a case of a 43-year-old male who presented with cardiorespiratory collapse as a result of life-threatening bronchial asthma that required 10 minutes of cardiopulmonary resuscitation. He was intubated and haemodynamically supported with inotropes for 4 days. Post-extubation, he developed progressively worsening action myoclonus involving limbs, trunk, and speech. His mini-mental state examination (MMSE) was 16/30. All his metabolic, infective, and neuroimaging screenings, including CT and MRI of the brain, were normal. He was diagnosed with LAS due to the classical onset and semiology. Clonazepam and sodium valproate were started, with piracetam added later. He showed marked functional improvement after the treatment. He was able to walk with walking-frame and MMSE improved to 26/30; however, minimal dysarthria persisted. Early and accurate diagnosis is of paramount importance for disability limitation; however, management of LAS can be challenging as high-quality, evidence-based treatment has not been established. This case highlights the importance of retaining high clinical suspicion in diagnosing LAS. Given its typical history and presentation, the diagnosis can be made confidently.
INTRODUCTION
It has been reported that in up to one-third of post resuscitated comatose patients experienced a seizure, with post-hypoxic myoclonus (PHM) as the most common type.1
PHM is defined as repetitive, generalised, focal or multifocal, myoclonic motor movements involving the face, limbs, or trunk that can occur at any time following cardiac arrest, and stems from increased neuronal excitability after brain injury.2
PHM can be classified by the time of onset, outcomes, response to treatment, characteristics of myoclonus, and neurological examination, with particular focus on the presence of coma. However, it is generally divided into acute PHM and chronic PHM or commonly known as Lance-Adams syndrome (LAS).
The author reports a case of LAS after a life-threatening bronchial asthma attack.
CASE REPORT
The author reports the case of a 43-year-old male who presented with life-threatening acute exacerbation of bronchial asthma precipitated by viral pneumonia. He had a case of partly controlled bronchial asthma on meter-dosed inhaler salbutamol and meter-dosed inhaler budesonide. On the day of presentation, he experienced worsening shortness of breath, purulent cough, and fever for 2 days.
He was brought to the emergency department with no sign of life and required 10 minutes of cardiopulmonary resuscitation. The patient was then intubated and haemodynamically supported with intravenous noradrenaline for 4 days in the intensive care unit. Post-extubation, he developed progressively worsening action myoclonus involving limbs, trunk, and speech. His mini-mental state examination (MMSE) was 16/30. All his metabolic, infective, and neuroimaging screenings, including CT (Figure 1) and MRI of the brain (Figure 2), were normal. Electroencephalogram (EEG) was not carried out due to the unavailability of the service. He was later diagnosed with LAS due to the classical onset of, and semiology of, myoclonus.
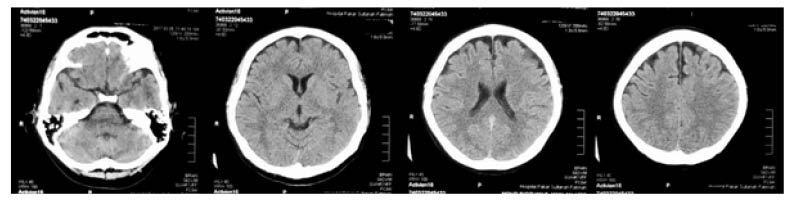
Figure 1: CT of the brain of a 43-year-old male with Lance-Adams syndrome.
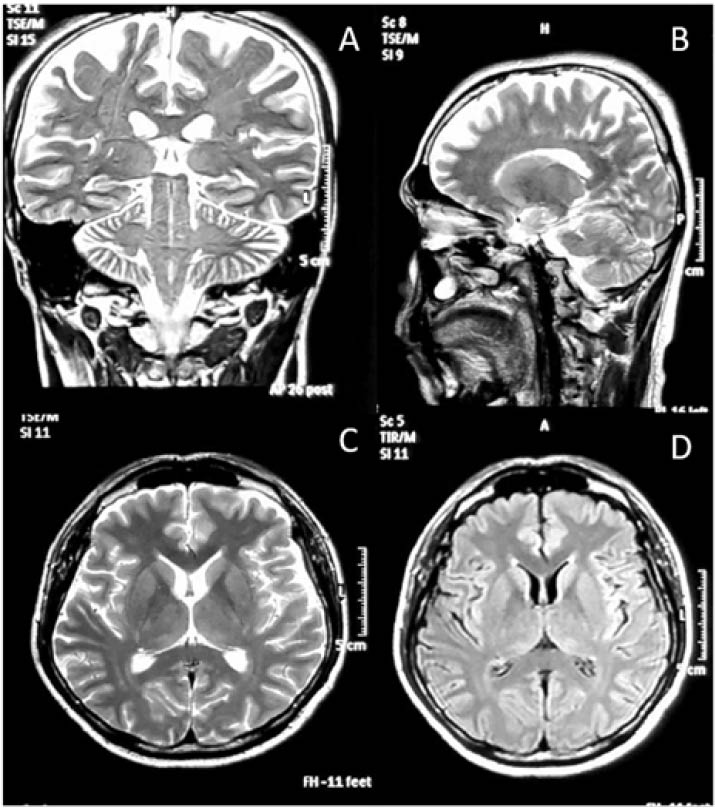
Figure 2: Magnetic resonance of the brain.
A) T2-weighted (coronal cut); B) T2-weighted (sagittal cut); C) T2-weighted (axial brain cut); D) FLAIR sequence.
A titrating dose of clonazepam was started, from 0.5 mg 3 times a day and titrated up to a total of 8.0 mg per day. The patient could not tolerate 8.0 mg of clonazepam because of drowsiness and lethargy. Clonazepam was then reduced to 6.0 mg a day with added sodium valproate. With clonazepam (6.0 mg/day) and sodium valproate (1.2 g/day), his action myoclonus was partially controlled. After reviewing multiple case reports, piracetam 2.4 g/day was initiated as an add-on therapy. He showed marked functional improvement after the combination of clonazepam (6.0 mg/day), sodium valproate (1.2 g/day), and piracetam (2.4 g/day). He was then able to ambulate with a walking frame, self-dress with minimal aid, and MMSE had improved to 26/30. However, minimal dysarthria persisted upon discharge after 2 weeks of admission. During clinic follow-up, sodium valproate was further titrated up to 1.8 g/day with aggressive physiotherapy and occupational therapy. To date, functionally, he can walk without help, write, and hold a spoon and fork for food, but residual action myoclonus with fine movement persisted; for example, he was not able to use a screwdriver due to action myoclonus. The outpatient EEG was normal.
DISCUSSION
Post hypoxic myoclonus is defined as repetitive sudden muscle contraction or muscle tone lapses that are brief, involuntary, and shock-like, involving face, limbs, or trunk, and can occur at any time following cardiac arrest due to increased neuronal excitability after brain injury.2,3 Multiple different classifications had been suggested, but it is predominantly divided into two main groups: acute PHM and chronic PHM. Reviews have suggested the importance of differentiating between acute and chronic PHM due to its prognostication value;2,4-6 however, in a clinical setting, it could be challenging to classify between different post cardiac arrests due to the extensive use of sedation as a neuroprotective measure. Freund and Kaplan2 suggested a summary of clinical features to differentiate between acute PHM and chronic PHM (Table 1).
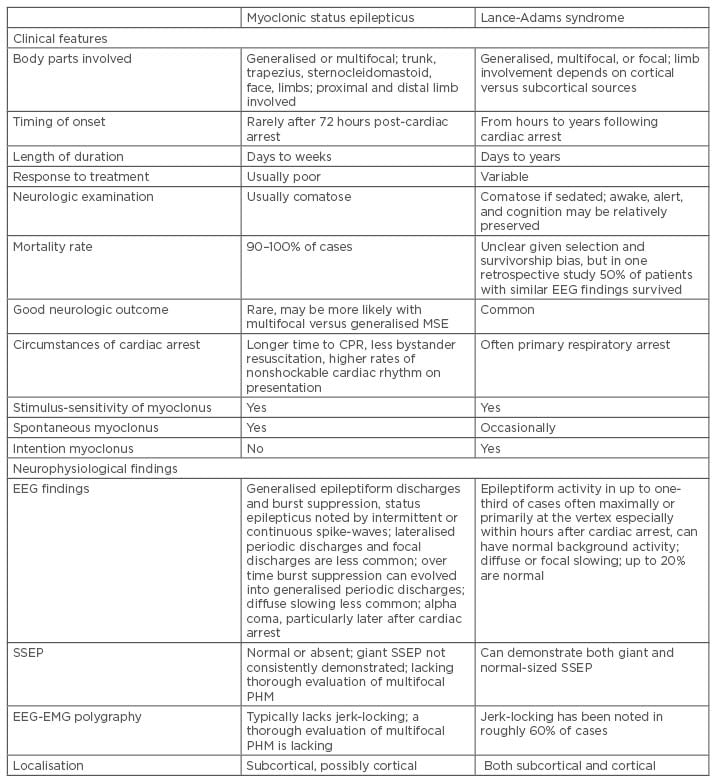
Table 1: Summary of clinical and electroencephalogram findings in post hypoxic myoclonus.2
CPR: cardiopulmonary resuscitation; EEG: electroencephalogram; EMG: electromyogram; PHM: post-hypoxic myoclonus; SSEP: somatosensory-evoked potential.
This case was consistent with the diagnosis of chronic PHM, also known as LAS. The syndrome was first reported by Lance and Adams in 1963 and typically, after recovery of consciousness, patients exhibit muscle jerks, which are at first generalised, but later become more restricted to upper or lower limbs, and are characteristically made worse by voluntary action.7 The severity of the myoclonus is proportionate to the precision of the task required and the patient may retain full consciousness during the episode of myoclonus. Other associated features include cerebral ataxia, dementia, spasticity, and incontinence.8 The most commonly reported adverse event associated with LAS was respiratory arrest.2 In this case, the patient showed classical onset and characteristic of action myoclonus post-cardiac arrest as a result of life-threatening bronchial asthma.
The pathophysiology of LAS is not entirely understood, although the literature has suggested that possible loss of serotonin (5-hydroxytryptamine or 5-HT) may be the cause of LAS.3,8,9 This was based on an earlier report that showed relatively low levels of serotonin activity in cerebrospinal fluid and partial response toward serotonergic treatment. However, the further autopsy report showed multiple different areas of intracranial abnormalities such as in mammillary bodies, brainstem raphe nuclei, cuneiform, subcuneiform nuclei, supratrochlear nucleus, lateral subnucleus of mesencephalic grey matter, and midbrain periaqueductal grey matter.2,3,6,7,9 The reason for multiple areas of abnormalities that have been suggested is different mechanisms of cardiac arrest involved.
Imaging studies in diagnosing LAS have not yielded satisfactory results. Both brain CT and brain MRI show no disease-specific abnormality; however, with the advancement of medical technologies, functional neuroimaging such as brain single-photon emission CT or brain PET could provide additional information about the anatomical and pathophysiological basis of LAS.10,11
Unlike neuroimaging studies, neurophysiological movement studies can provide more useful information in guiding the decision making of the diagnosis and treatment modality. LAS typically shows focal EEG discharge over the sensorimotor cortical area, especially when using back-averaging of the EEG by jerk-locking or electromyogram discharge during the acute phase.2,3 This could explain the semiology of action-induced myoclonus in LAS. Additionally, by identifying foci of myoclonus in EEG, area-specific medication can be prescribed more confidently. In this case, neurophysiological movement studies were not performed in the acute phase owing to unavailability of service, but subsequent EEG study was normal.
Given how rare this disease is and the lack of understanding of its exact pathophysiology, the treatment regime for LAS is frequently based on case reports or case series. Benzodiazepines, clonazepam, in particular, a chlorinated derivative of nitrazepam that was first introduced into clinical practice in 1966, has been used successfully to treat LAS since the 1970s.12,13 It facilitates γ-Aminobutyric acid (GABA) transmission in the brain by directly affecting benzodiazepine receptors. Benzodiazepines do not affect 5-HT use in the brain and block the egress of 5-Hydroxyindoleacetic acid from the brain; however, these effects contradict the beneficial effect of 5-HT in human myoclonic features. It was postulated that the possibility of its benefit lies in benzodiazepine receptors.14 Nevertheless, it is considered the first-line treatment in LAS. As LAS is associated with cortical origin myoclonus, the sodium valproate used has achieved a certain degree of success in treating LAS with the postulating mechanism through the elevation of brain GABA level in synaptic regions.3 However, due to the side effects of sodium valproate such as tiredness, weight gain, tremor, and hair loss, it may render a higher dosage intolerable, similarly to the patient in the case reported herein.15 Piracetam, a derivative of GABA, which postulated the effect of improved neuroplasticity, has shown to be beneficial in controlling myoclonus.16 Recent studies also reported the efficacy of levetiracetam in treating LAS.17,18 Clonazepam, sodium valproate, piracetam, and levetiracetam are the first-line therapy options in the management of LAS. Combination treatment may achieve the best possible outcome in these patients.
The prognosis had been reported to be good in LAS if early aggressive rehabilitation and appropriate drug treatment initiated.3,5,6,9,15,18
CONCLUSION
Early recognition and accurate diagnosis of LAS is of paramount importance to correctly predict post hypoxic myoclonus in the patient, to decide on the most effective treatment regimen, and for disability limitation. However, management of LAS can be challenging as high-quality, evidence-based treatment has not been established. This case report highlights the importance of retaining high clinical suspicion in diagnosing LAS. Given its typical history and presentation, the diagnosis can be made confidently.