Chairperson: Fiona Blackhall1,2
Speakers: Fabrice Barlesi,3 Maximilian Hochmair,4 Rosario García Campelo5
1. The Christie NHS Foundation Trust, Manchester, UK
2. The University of Manchester, Manchester, UK
3. Institut Gustave Roussy, Paris, France
4. Karl Landsteiner Institute of Lung Research and Pulmonary Oncology, Vienna, Austria
5. University Hospital A Coruña, A Coruña, Spain
Disclosure: The speakers who participated in this symposium received honoraria from Takeda Oncology. Prof Blackhall has received fees for advisory board attendance, consultancy, or speaker honoraria from AbbVie, AstraZeneca, Bayer, Boehringer Ingelheim, Janssen, MSD, Pfizer, Roche, and Takeda. Prof Barlesi has reported personal financial interests with AstraZeneca, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, Eli Lilly Oncology, F. Hoffmann-La Roche Ltd, Novartis, Merck, MSD, Pierre Fabre, Pfizer, and Takeda; institutional financial interests with AbbVie, ACEA, Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Boehringer Ingelheim, Eisai, Eli Lilly Oncology, F. Hoffmann-La Roche Ltd, Genentech, Ipsen, Ignyta, Innate Pharma, Loxo, Novartis, Medimmune, Merck, MSD, Pierre Fabre, Pfizer, Sanofi-Aventis, and Takeda; and has served as the principal investigator for trials sponsored by AstraZeneca, Bristol Myers Squibb, Merck, Pierre Fabre, and F. Hoffmann-La Roche Ltd. Dr Hochmair has received fees for advisory board attendance and speaker honoraria from Bayer, Bristol Myers Squibb, Boehringer Ingelheim, MSD, Roche, and Takeda. Dr García Campelo has received fees for advisory board attendance, consultancy, and speaker honoraria from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Lilly, MSD, Novartis, Roche, and Takeda.
Acknowledgements: Writing assistance was provided by Dr Laura Harrison, Excel Medical Affairs, Fairfield, Connecticut, USA, and was funded by Takeda Oncology.
Support: The publication of this article was funded by Takeda Oncology. The views and opinions expressed are exclusively those of the speakers.
Citation: EMJ Oncol. 2020;8[1]:33-42.
Meeting Summary
Despite significant advancements in recent years, lung cancer remains the leading cause of cancer-related deaths globally. The promise of precision medicine in non-small cell lung cancer (NSCLC) is starting to become a reality owing to the introduction of numerous tyrosine kinase inhibitors targeting specific oncogenic alterations. Therefore, there is an even greater need for accurate, rapid, and accessible testing to allow for large-scale molecular profiling of patients with NSCLC. The evolution of the treatment landscape for patients with NSCLC harbouring an anaplastic lymphoma kinase (ALK) gene rearrangement provides an excellent example of the impact of targeted therapy. Four different ALK inhibitors are now recommended by clinical practice guidelines for the first-line treatment of ALK-positive metastatic NSCLC: crizotinib, ceritinib, alectinib, and brigatinib. However, despite demonstrating significant improvements in progression-free survival (PFS), disease progression and relapse in patients with advanced NSCLC is inevitable. In addition, the occurrence of brain metastases is common in patients with advanced ALK-positive NSCLC, and penetration of the blood–brain barrier by ALK inhibitors is important to achieve the best possible patient outcomes. Selecting the right therapy and sequencing treatments appropriately is essential to ensure each patient receives the optimal treatment for them.
The objective of this satellite symposium held at the European Society for Medical Oncology (ESMO) Virtual Congress was to provide an educational forum to discuss key concepts associated with testing and first-line treatment strategies in NSCLC, with a specific focus on ALK-positive disease, in order to emphasise the importance of providing truly personalised patient care.
Introduction
Professor Fiona Blackhall
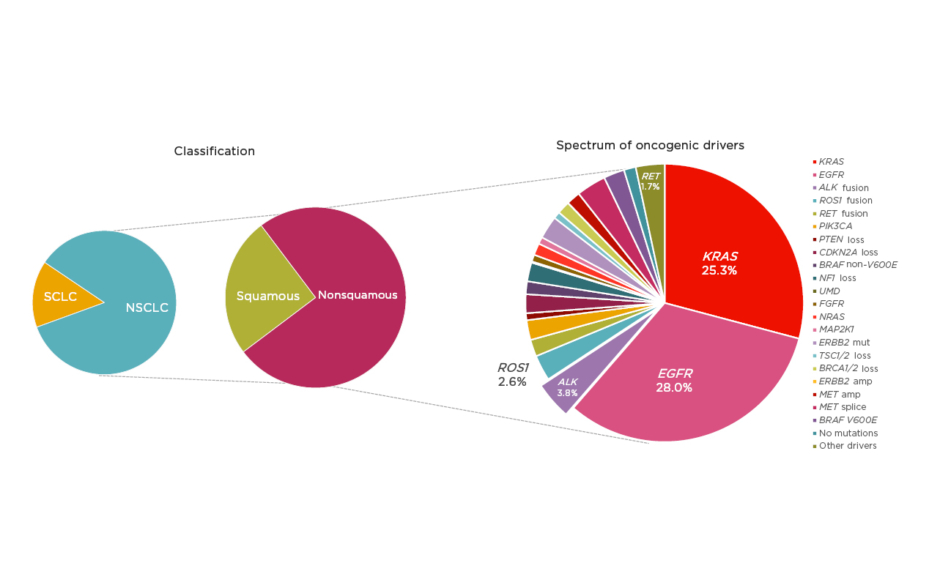
The oncology community has been experiencing an unprecedented moment in lung cancer diagnosis and treatment; however, despite significant advances, lung cancer remains the leading cause of cancer mortality worldwide.1 NSCLC, accounting for approximately 85% of lung cancer cases,2 provides a pivotal example of how appropriate disease segmentation and treatment personalisation can markedly impact patient outcomes. NSCLC can be classified as squamous cell carcinoma (30%) or nonsquamous carcinoma (70%).2 Substantial progress in the understanding of the disease in recent years has led to further subclassification of nonsquamous NSCLC into various molecular subtypes according to specific oncogenic driver mutations or gene translocations (Figure 1).3,4 As a result, there is a growing list of targeted therapies that can be used to treat specific subsets of patients, including those with mutations in the epidermal growth factor receptor (EGFR) gene, or translocations in the ALK or c-ros oncogene 1 (ROS1) genes.
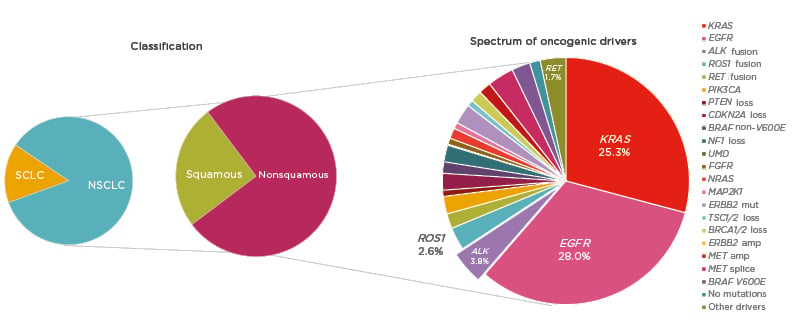
Figure 1: Subclassification of non-small cell lung cancer.
amp: amplification; mut: mutation; NSCLC: non-small cell lung cancer; SCLC: small cell lung cancer.
Adapted from Jordan et al.4
Mutations in EGFR are present in approximately 15–30% of patients with NSCLC,2,4 and classical activating mutations, such as EGFR exon 19 deletions and point mutations in exon 21 (L858R), are associated with responsiveness to targeted tyrosine kinase inhibitor (TKI) treatment.5 In addition, many of the less commonly observed alterations, such as EGFR exon 19 insertions and point mutations in exon 21 (L861Q), exon 18 (G719X), and exon 20 (S768I), are also responsive to TKI therapy.5 However, exon 20 insertions are associated with poorer TKI responses.6 Future research aims to further characterise these additional alterations, and three treatments are currently under investigation for patients with EGFR exon 20 insertions (mobocertinib, amivantamab, and poziotinib).
ALK rearrangements are identified in 3–5% of patients with NSCLC and are usually mutually exclusive with EGFR mutations or ROS1 rearrangements.3,4,7 Echinoderm microtubule-associated protein like-4 (EML4) is a common fusion partner of ALK and there are multiple EML4–ALK variants.8 Variant status is associated with clinical outcome and may have implications for specific treatment strategies.9 Patients with ALK-positive NSCLC are more likely to have adenocarcinoma histology and to be never smokers.10
Over the last decade, several ALK inhibitors have been developed for the first-line treatment of ALK-positive NSCLC, including the first-generation inhibitor crizotinib and the second-generation inhibitors ceritinib, alectinib, and brigatinib. Lorlatinib was the first third-generation inhibitor to be approved for ALK-positive NSCLC, and several inhibitors are also in development, including ensartinib and entrectinib.
With the availability of potent targeted treatments, it is increasingly important to ensure upfront molecular testing is performed to identify oncogenic drivers. However, there is some evidence to suggest that, despite improvements, real-world ALK testing rates remain suboptimal.11 More work is also needed to ensure specificity and tolerability of first-line treatments to suit individual patient needs. Disease progression in NSCLC is inevitable and clinicians must consider the optimal treatment sequencing strategy to achieve the best outcomes for patients. In addition, the central nervous system (CNS) is a known sanctuary site for NSCLC and brain metastases occur frequently in advanced ALK-positive disease.12 Improved penetration of second-generation ALK inhibitors into the CNS may improve outcomes for these patients.13
Oncogenic Driver Testing Strategies: Identifying the Right Patients for the Right Treatment
Professor Fabrice Barlesi
Advancements in high-throughput technologies over the past decade have led to a rapid reduction in the costs associated with genome sequencing,14 allowing these techniques to become more globally accessible. As an increasing number of targetable molecular alterations in NSCLC are identified, it is more important than ever to ensure that patients with advanced NSCLC are accurately genotyped.
The results of the 1-year nationwide French Cooperative Thoracic Intergroup (IFCT) study conducted in 2012 clearly demonstrated that the identification of actionable targets through molecular profiling provided a clinical benefit.15 The presence of a genetic alteration was associated with improved overall survival (16.5 months; 95% confidence interval [CI]: 15.0–18.3) compared with the absence of a genetic alteration (11.8 months; 95% CI: 10.1–13.5) (Figure 2).15 In addition, the results of the MOSCATO 01 trial16 in patients with advanced disease refractory to standard treatment showed that matching patients to targeted therapy using high-throughput genomic analyses was associated with improved PFS.16
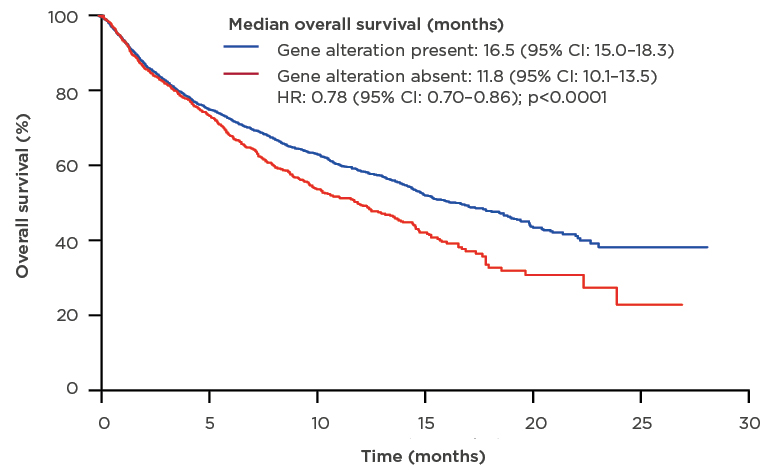
Figure 2: Median overall survival of patients who underwent molecular analysis for genomic alterations.
CI: confidence interval; HR: hazard ratio.
Adapted from Barlesi et al.15
Guidelines for metastatic NSCLC, including the American Society of Clinical Oncology (ASCO)/International Association for the Study of Lung Cancer (IASLC) molecular testing guidelines and the ESMO clinical practice guidelines, clearly highlight the need for testing molecular alterations associated with targeted therapies approved by the U.S. Food and Drug Administration (FDA). These include ALK, EGFR, ROS1, and v-raf murine sarcoma viral oncogene homolog B1 (BRAF).17,18 The recent National Comprehensive Cancer Network (NCCN) guidelines also recommend testing for mesenchymal epithelial transition factor (MET) exon 14 skipping and rearranged during transfection (RET) alterations.19
There are a wide range of possible testing solutions to identify molecular alterations. Immunohistochemistry and fluorescence in situ hybridisation are both approved and widely utilised methods for testing ALK and ROS1 rearrangements. The advent of high-throughput next-generation sequencing (NGS), which allows simultaneous assessment of multiple genes, has the potential to revolutionise the field. The French National Cancer Institute (INCa) has supported the implementation of a national network of 28 hospital molecular genetics platforms. Data from this network show that >18,000 patients were screened using an NGS panel in 2017.20 Commercially available genomic profiling solutions are also available, including the tissue-based FoundationOne® CDx (Foundation Medicine, Cambridge, Massachusetts, USA), which can detect >300 gene mutations as well as selected gene rearrangements, including EGFR and ALK.21 The French Plan for Genomic Medicine 2025 (Inserm, Paris, France), launched in 2016, includes two high-throughput sequencing platforms aiming to offer centralised and efficient whole-exome sequencing data to clinicians. This may provide important data for those patients in whom alterations cannot be identified using commercial solutions.
Choosing the best testing technique can be a complex process with multiple determinants. Turnaround time is an important factor in patients with advanced disease. The results from immunohistochemistry can be available within 48 hours, whereas NGS may take up to 1–2 weeks,22,23 although timing may be region-dependent. The cost of the test also plays an important role. Commercially available solutions may be more expensive; however, the number of alterations that need to be tested is a key consideration. Several studies have demonstrated that upfront NGS is a more cost-effective method when testing for multiple molecular alterations compared with sequential testing strategies.24,25 Several randomised studies are currently ongoing, including the SAFIR02 and PROFILER 02 studies, to investigate the added value of a large molecular profiling panel compared with a more limited panel. It is important to consider the availability of therapeutic solutions to target drivers and to keep in mind that the presence of a target does not necessarily lead to targeted treatment. In the MOSCATO 01 study, only 19% of patients went on to receive a targeted therapy.16 The approval and reimbursement status of treatments is also a key factor, although certain investigational products may be available through expanded access programmes or clinical trials.
The site of tissue acquisition is also significant. Testing for genomic alterations in cell-free DNA (cfDNA) from blood samples presents a more practical option for patients than tissue sampling and may enable clinicians to offer more effective personalised treatments. Commercial liquid biopsy tests are available, including FoundationOne Liquid (Foundation Medicine) and Guardant360® (Guardant Health, Redwood City, California, USA), which can analyse >70 genomic alterations in blood samples.22,26 Noninferiority of comprehensive cfDNA testing compared with tissue genotyping in patients with advanced NSCLC has been demonstrated, with cfDNA showing >98% concordance.27 In addition, liquid biopsy had a faster turnaround time of 9 days compared with 15 days for tissue genotyping.27 BFAST28 was the first prospective study to demonstrate the clinical utility of blood-based NGS testing to identify patients who were ALK-positive and select targeted therapy.28 This raises the potential for NGS from liquid biopsy samples as the future for testing in patients with NSCLC.
Treatment Selection in ALK-Positive Metastatic NSCLC: Optimising Outcomes
Doctor Maximilian Hochmair
Historically, the treatment of lung cancer was simple, owing to limited treatment options; however, the survival rate for patients was low. An increasing number of targeted treatment options are now available or under investigation, including for patients with molecular alterations in EGFR, ROS1, BRAF, NTRK, MET, HER2, and RET. This has led to a paradigm shift away from chemotherapy as the primary first-line treatment option for patients with NSCLC. As the number of options available for treating patients with ALK-positive NSCLC increases, treatment selection, strategy, and sequencing to optimise patient outcomes are of primary importance.
Personalisation of treatment in patients with ALK-positive NSCLC is vital. ALK rearrangements are associated with a lower response to immune checkpoint inhibitors. A retrospective analysis of 58 patients demonstrated an objective response rate (ORR) of 3.6% in patients with EGFR-mutated or ALK-positive NSCLC treated with programmed cell death protein 1 or programmed death-ligand 1 inhibitors, compared with an ORR of 23.3% for patients with EGFR wild-type or ALK-negative NSCLC (p=0.053).29 Therefore, it is important to wait for oncogenic testing results before starting first-line treatment. Four different ALK inhibitors are now recommended by the ESMO and NCCN clinical practice guidelines for first-line treatment of ALK-positive metastatic NSCLC: crizotinib, ceritinib, alectinib, and brigatinib.18,19
Crizotinib was the first ALK inhibitor approved for the treatment of ALK-positive NSCLC, and the PROFILE 1014 trial30 was the first Phase III study to demonstrate the efficacy of an ALK inhibitor compared with chemotherapy in the first-line setting. Median PFS for crizotinib, assessed by the Independent Review Committee (IRC), was 10.9 months (95% CI: 8.3–13.9) compared with 7.0 months (95% CI: 6.8–8.2) for chemotherapy. Crizotinib-associated adverse events (AE) included vision disorders, diarrhoea, nausea, and oedema.30
Subsequently, several second-generation ALK inhibitors have been developed. The ASCEND-4 Phase III trial31 comparing ceritinib with chemotherapy demonstrated a median IRC-assessed PFS of 16.6 months (95% CI: 12.6–27.2) for ceritinib and 8.1 months (95% CI: 5.8–11.1) for chemotherapy. Ceritinib was associated with gastrointestinal side effects, including nausea and diarrhoea.31 The ALEX32,33 and ALTA-1L34 trials provided a head-to-head comparison of the first-generation crizotinib with the second-generation inhibitors alectinib and brigatinib, respectively. The ALEX trial showed a median IRC-assessed PFS of 25.7 months (95% CI: 19.9–not reached [NR]) for alectinib compared with 10.4 months (95% CI: 7.7–14.6) for crizotinib32 (investigator-assessed PFS with alectinib was 34.8 months [95% CI: 17.7–NR] and with crizotinib was 10.9 months [95% CI: 9.1–12.9])33 in patients with no prior treatment for advanced disease.32 Relevant alectinib-related AE included liver enzyme elevation and myalgia.33 In ALTA-1L, the median IRC-assessed PFS for brigatinib was 24.0 months (95% CI: 18.5–NR) compared with 11.0 months (95% CI: 9.2–12.9) for crizotinib (investigator-assessed PFS with brigatinib was 29.4 months [95% CI: 21.2–NR] and with crizotinib was 9.2 months [95% CI: 7.4–12.9]) in patients with no prior ALK inhibitor treatment and, at most, one prior systemic therapy.34 Brigatinib-associated AE included increased creatine kinase levels, cough, and hypertension. Exploratory analyses from the ALTA-1L trial also evaluated the impact of EML4–ALK fusion variant status on the clinical efficacy of brigatinib compared with crizotinib. These analyses found that patients with EML4–ALK variant 3 had worse PFS regardless of treatment. Brigatinib was associated with superior PFS to crizotinib regardless of ALK fusion variant status.9 Data from the eXalt3 randomised Phase III35 trial of ensartinib were recently presented at the IASLC World Conference on Lung Cancer (WCLC), demonstrating a median IRC-assessed PFS of 25.8 months (95% CI: 21.8–NR) for ensartinib compared with 12.7 months (95% CI: 9.2–6.6) for crizotinib. Low-grade rash and transaminitis were the most frequent ensartinib-related AE.35
Unfortunately, relapse and disease progression in patients with advanced NSCLC on targeted therapy is unavoidable owing to the development of ALK resistance mutations or bypass signaling.36 Rebiopsy of tissue following ALK inhibitor failure is an option, although guidelines do not currently recommend this as mandatory for treatment decisions.18,37 The frequency and range of ALK resistance mutations differs depending on the specific ALK inhibitor.36 The third-generation ALK inhibitor lorlatinib has been shown to have strong efficacy in patients who have received prior treatment with a second-generation ALK inhibitor,38 and the presence of ALK resistance mutations has been shown to be associated with sensitivity to lorlatinib in patient-derived cell lines.36 In contrast, cell lines without ALK resistance mutations were resistant to lorlatinib. This may allow clinicians to personalise treatment sequencing strategies on the basis of a specific patient’s ALK resistance mutation status.36 Following this symposium, results of the Phase III CROWN trial,39 comparing lorlatinib with crizotinib in first-line treatment, were presented at ESMO 2020. These data showed a 72% improvement in IRC-assessed PFS in patients treated with lorlatinib compared with crizotinib (hazard ratio [HR]: 0.28), with a median follow-up for PFS of 18.3 months (95% CI: 16.4–20.1) for lorlatinib and 14.8 months (95% CI: 12.8–18.4) for crizotinib. The majority of lorlatinib-related AE were laboratory abnormalities.39
Reaching the Sanctuary Site: Options for Patients with ALK-Positive NSCLC with Brain Metastases
Doctor Rosario García Campelo
The introduction of the first-generation ALK inhibitor crizotinib changed the treatment paradigm in ALK-positive NSCLC. The ALEX, ALTA-1L, and eXalt3 trials have all shown improved median PFS and duration of response for second-generation ALK inhibitors compared with first-generation treatment (Figure 3).30-35,40-43 However, the site of disease metastases continues to be an important factor when making treatment decisions for these patients.
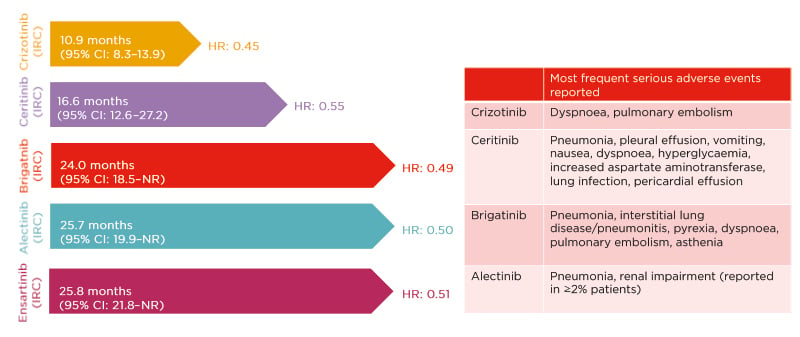
Figure 3: Efficacy and safety of anaplastic lymphoma kinase inhibitors in the first-line setting.30-35,40-43
Unadjusted, indirect comparison for illustration only; clinical significance is not implied. Cross-trial comparisons are potentially confounded by differences in trial design and study population.
CI: confidence interval; HR: hazard ratio; IRC: Independent Review Committee; NR: not reached.
The occurrence of CNS metastases is common in patients with advanced NSCLC, and approximately 30% of patients with ALK-positive Stage IV NSCLC have brain metastases at baseline.44 In addition, patients with ALK-positive NSCLC are at higher risk of developing brain metastases during the course of disease.45 The cumulative incidence of brain metastases was shown to be significantly higher for ALK-positive NSCLC compared with ROS1-positive cancers (p=0.0039).46 Brain metastases also have a negative impact on patient quality of life, with patients with NSCLC and brain metastases reporting a significantly lower general health status questionnaire (EQ-5D) score (0.52; p≤0.05) than patients with metastases at other sites, including the liver (0.71) and adrenal glands (0.83).47 Management of ALK-positive NSCLC in patients with CNS metastases is also associated with higher costs. A recent study showed an increase in annual costs of approximately €15,000 compared with the management of patients without CNS metastases.48 Whole brain radiotherapy (WBRT) is often used to treat patients with NSCLC and symptomatic brain metastases. However, several randomised clinical trials have suggested that WBRT may be associated with cognitive decline.49-51 Incorporating ALK inhibitors into first-line treatment may allow WBRT to be postponed, deferring potential long-term neurocognitive impairment to later in the disease course.
The concentration of crizotinib is lower in cerebrospinal fluid than in plasma. In a case example from a patient with ALK-positive NSCLC with intracranial progression, the cerebrospinal fluid-to-plasma crizotinib ratio was 0.0026, signifying poor blood–brain barrier penetration. This allows the brain to act as a ‘sanctuary site’ for tumour growth.13,52 Novel second-generation ALK inhibitors have the potential for improved CNS efficacy. Results from the ALEX study demonstrated an ORR of 81% (95% CI: 58–95) for alectinib compared with 50% (95% CI: 28–72) for crizotinib in patients with measurable CNS lesions at baseline.32 The median investigator-assessed PFS in patients with CNS metastases at baseline was 25.4 months for alectinib compared with 7.4 months for crizotinib, and the HR for PFS with any brain metastases was 0.37.53 In the ALTA-1L trial the confirmed intracranial ORR was 78% (95% CI: 52–94) for brigatinib compared with 26% (95% CI: 10–48) for crizotinib. The median IRC-assessed PFS in patients with any brain metastases at baseline was 24.0 months (95% CI: 18.4–NR) for brigatinib compared with 5.6 months (95% CI: 3.8–9.4) for crizotinib. The HR for PFS with any brain metastases was 0.25.34 There are currently limited data from the eXalt3 study on the impact of ensartinib on brain metastases; however, initial results indicated an IRC-assessed confirmed intracranial ORR of 64% for patients treated with ensartinib compared with 21% for patients treated with crizotinib.35 Recent data presented from the CROWN study showed an IRC-assessed intracranial ORR of 82% (95% CI: 57–96) for lorlatinib compared with 23% (95% CI: 5–54) for crizotinib in patients with measurable brain metastases at baseline.39 These data indicated that there was a clear benefit of using second- or third-generation ALK inhibitors in patients with brain metastases.
Patients with NSCLC are often highly symptomatic54 and are exposed to targeted therapy-associated toxicities over long periods of time.55 Patient-reported outcome measures provide a more direct method of reporting outcomes that are relevant to the patient, including symptom and treatment burden and health-related quality of life (HRQoL).56 Several clinical trials have reported data on patient HRQoL. During the ALEX trial, no significant difference in the European Organisation for Research and Treatment of Cancer (EORTC) core QoL questionnaire (QLQ-C30) score or the lung cancer supplement to the QLQ-C30 (QLQ-LC13) score was reported for patients treated with alectinib compared with those treated with crizotinib.57 Conversely, the ALTA-1L trial demonstrated a significant improvement in HRQoL for patients treated with brigatinib compared with crizotinib. The median time-to-worsening HRQoL was 26.7 months for patients treated with brigatinib compared with 8.3 months for those treated with crizotinib, and brigatinib significantly prolonged the duration of improvement in HRQoL (p<0.001) compared with crizotinib. In addition, brigatinib demonstrated a numerical improvement in all functional domains, with substantial improvement in cognitive functioning scores (estimated difference: 4.9 [95% CI: 1.7–8.1]).34,58 Targeted precision-based treatment for patients with advanced NSCLC has come a long way over the last decade. However, there are still a number of challenges that need to be overcome to continue improving patient outcomes and to enable the delivery of truly personalised care.
Conclusions
Upfront testing for molecular alterations is essential to ensure personalised treatment for patients with NSCLC, and NGS, which provides a cost-effective solution for analysing a large number of genes in a single panel, should become the widely adopted standard. Various effective ALK inhibitors are recommended for the first-line treatment of ALK-positive metastatic NSCLC, including crizotinib, ceritinib, alectinib, and brigatinib. However, multiple mechanisms ultimately drive resistance to ALK inhibitors, leading to disease progression. Some treatments may be able to overcome specific resistance mutations; however, rebiopsy at progression may be required to help guide treatment decisions in evolving disease. CNS metastases are frequent in patients with ALK-positive NSCLC and are associated with a negative prognosis and poorer HRQoL. Poor penetration of the blood–brain barrier by crizotinib may allow the CNS to act as a sanctuary site for tumour growth. Second-generation ALK inhibitors are a potent first-line treatment option for patients with ALK-positive NSCLC with brain metastases. Despite numerous advances in available therapeutics, optimal treatment sequencing remains an unmet need in patients with ALK-positive NSCLC, and the availability of third-generation ALK inhibitors may impact this.






