
Interviewee: Rianne Oostenbrink
ENCORE – NF1 expertise center, Erasmus MC, Rotterdam, The Netherlands
Disclosure: Oostenbrink is an advisory consultant for AstraZeneca, a member of the European Union (EU) Patient-centric clinical trial platform (EU-PEARL), and a Full Member of the European Reference Network on Genetic Tumor Risk Syndromes (ERN GENTURIS). EU-PEARL has received funding from the Innovative Medicines Initiative (IMI) 2 Joint Undertaking under grant agreement No. 853966. This Joint Undertaking receives support from the EU’s Horizon 2020 research and innovation programme and European Federation of Pharmaceutical Industries and Associations (EFPIA) and Children’s Tumor Foundation, Global Alliance for TB Drug Development non-profit organisation, and Springworks Therapeutics Inc. This publication reflects the authors’ views. Neither IMI nor the EU, EFPIA, or any Associated Partners are responsible for any use that may be made of the information contained herein.
Acknowledgements: Writing assistance was provided by Eleanor Roberts, Beeline Science Communications, London, UK.
Support: This article was supported by an educational grant from AstraZeneca.
Disclaimer: The opinions expressed in this article belong solely to the named interviewee.
Citation: EMJ. 2021;6[4]:26-31.
Summary
Neurofibromatosis Type 1 (NF1) is a genetic disorder, generally diagnosed during early childhood, that affects around 1 in 3,000 people worldwide. Around 30−50% of patients with NF1 develop NF1-associated plexiform neurofibromas (PN). These benign tumours, located on peripheral nerve sheaths, carry a lifelong risk for malignancy of 8−13%. PNs first present in childhood and, depending on their size and location, may cause pain, organ compression, disfigurement, and other complications. Though rare, the incidence is high enough that every general practitioner (GP) should expect to see at least one patient with NF1 in their practice. As such, it is important for all healthcare professionals (HCP) to understand the defining signs of NF1 and PNs, so as not to misdiagnose, and fail to refer or treat the patient in specialised centres, with dedicated multidisciplinary teams (MDT). Rianne Oostenbrink, Associate Professor and Paediatrician at Erasmus Medical Center in Rotterdam, the Netherlands, spoke to EMJ about the current diagnostic pathway for people with PNs, the burden of disease, and the HCPs involved in patient care. She also highlighted the presence and work of the Europe-wide organisations that can support HCPs, and patients alike.
INTRODUCTION
NF1 is an autosomal dominant disorder, arising from a mutation in the gene that encodes neurofibromin. It is a tumour-predisposing condition, associated with many different manifestations, including the risk of developing benign tumours in the peripheral and central nervous systems. PNs, one complication of NF1, are benign, diffuse tumours, located on peripheral nerve sheaths, that may be visible from the outside, or present internally (Figure 1). Other manifestations of NF1 include café-au-lait macules (CALM); cutaneous neurofibromas; Lisch nodules in the iris; axillary- or inguinal-area freckling; skeletal dysplasias; behavioural and cognitive deficits; low-grade gliomas; and organ involvement.2
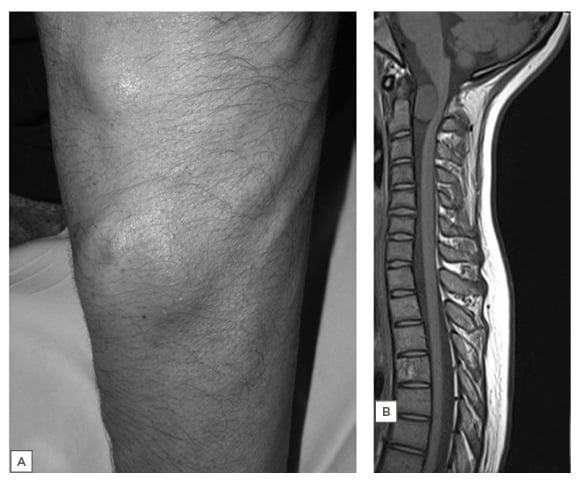
Figure 1: Plexiform neurofibroma in the forearm.
A) Subcutaneous NF in the upper limb in a patient with NF1. B) T1 sagittal MRI in an asymptomatic patient with C2 NF causing spinal cord flattening and distortion.
Adapted from Ferner et al. (2013).1
NF: neurofibromatosis; NF1: neurofibromatosis Type 1.
NF1 occurs in around 1 in 3,000−4,000 people,3 and around 30−50% with NF1 develop PNs.4,5 In general, growth rate is higher at younger ages, although growth can be variable at any age.6 Although some mutations are related with higher PN rates, for most patients, reported Oostenbrink, whether or not PNs will develop cannot be predicted based on other NF1 symptoms, such as the number of CALMs.
MEDICAL AND PSYCHOSOCIAL BURDENS ASSOCIATED WITH PLEXIFORM NEUROFIBROMAS
“The complexity of NF1,” Oostenbrink explained, “is that it affects many body systems in variable
ways and times. It impacts on physical functioning and cognition and development combined. The burden of PNs,” she continued, “is very variable. This can arise from PN visibility, size, and impact on functioning, but they may also have a large interindividual perception.”
PNs can cause pain and disfigurement, particularly in older children, adolescents, or adults, due to nerve or surrounding tissue compression, bone erosion, or organ displacement.6 NF1 can also cause fatigue, function loss, cognitive problems, and sleep difficulties, as well as burdens associated with stigma, appearance, social activity limitations and, later in life, career, and relationships.6
Another PN-associated burden Oostenbrink discussed is the lifelong need for medical monitoring, clinical investigations, and, where necessary, treatment; for instance, the time burden of an MRI that, for young children, is carried out under anaesthesia. Though not common (8−13%), there is a risk that PNs will develop into a malignant tumour,2,6 and a further burden is the worry this can cause. This can be difficult for both the patient, and their parents or caregivers, to cope with.
With all of this in mind, the burden of PNs should always be considered when supporting patients throughout their life.
DIAGNOSIS, TREATMENT, AND MONITORING OF NEUROFIBROMATOSIS TYPE 1 IN PATIENTS WITH PLEXIFORM NEUROFIBROMAS
While the incidence of NF1 is low, Oostenbrink discussed how it is known as the ‘largest small disease’. As there is roughly one NF1 patient for each GP, she underlined the importance of creating awareness at this level. One of the biggest barriers that HCPs face in recognising NF1 or PNs, especially for those with little prior experience of this condition, is in differentiating NF1 and PNs from other, similar, benign tumour-producing conditions such as lipoma (Table 1).
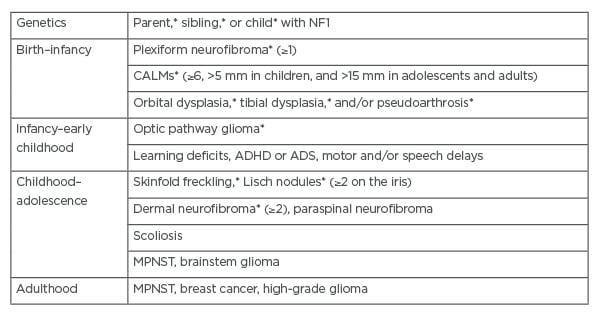
Table 1: Clinical features of neurofibromatosis Type 1.
*Diagnosis of NF1 requires two or more of the criteria, or one of the criteria and a proven genetic pathogenic mutation in the NF1 gene.
Adapted from Gutmann et al. (2017)1 and Brosseau et al. (2020).7
ADHD: attention deficit and hyperactivity disorder; ASD: autism spectrum disorder; CALM: café-au-lait macules; MPNST: malignant peripheral nerve sheath tumours.
“Checking skin is part of general training,” said Oostenbrink, “but some manifestations should lead to a specific diagnosis. CALMs are quite well known, but if a patient comes in just with a benign tumour in the skin, and CALMs are not that marked, or the main problem is in the leg, and the HCP doesn’t think about looking under the shirt for CALMs, they might not recognise it.” Just over half of people with NF1 have an inherited mutation,8 so children of parents or siblings with NF1 may be checked more routinely.
If NF1 or PNs are suspected, patients must be referred to a specialist centre with an MDT. This should involve those skilled in diagnosing and monitoring PNs, and understanding their cause, working alongside specialist surgeons, and oncologists, depending on tumour type; and, if needed, other specialists such as an orthopaedic team and a pain management team. Together, they can help explain to the patient and caregivers about PNs, and discuss potential treatment options: for example, surgery versus systemic treatment or symptom management.
As the manifestation of PNs may be diverse, treatment, discussed Oostenbrink, requires a multidisciplinary approach. This must be based around understanding the spectrum of PNs, what is important to the patient, and what their outcome expectations are. Treatment is most often with surgery, although drug treatments, such with the mitogen-activated protein kinase inhibitor selumetinib, are becoming available for cases where tumours are inoperable and symptomatic.7 Surgery may result in complete or partial removal, depending on specific circumstances, nerve and tissue involvement, and location. However, problems encountered with surgery can include large blood loss, nerve injury, disfigurement, and regrowth.9
Due to individual patient differences, even the same tumour in two people may need a different approach. The decision about treatment should be made with knowledge of the underlying condition, and its expected course. This is vital, because if the HCP knows it is a PN, says Oostenbrink, they then know there are some approaches that will not help. An example of where this was not carried out, she recalled, was the case of a child who underwent repeated surgery for a facial tumour that kept returning. On later presentation at an NF centre, the immediate recognition of CALMs helped to confirm the PN diagnosis and guide proper treatment.
Although the patient pathway may be different in different countries, one common feature is the need to have each patient evaluated by a dedicated and expert MDT. Once evaluated at a specialist centre, the result may be either further investigations, an intervention, or a referral back to a regional centre for monitoring. A patient may then return to the specialist centre after a defined time interval, if there is a change in PN, or other NF1 manifestations. To aid in this model, Oostenbrink described the development in the Netherlands of a national network, arising from her expertise centre, as nationwide as the number of patients was too high for one centre to manage. This network fulfils the need for patients to be monitored close to home if possible, and for care to be more centralised when necessary.
Once diagnosed with PN, Oostenbrink described how it will usually be the patient who picks up on a change in pain or growth. “Our role then is to discuss with the patient, and to make it more objective [when assessing] whether it is indeed a change in substantial growth, or clinical function. The key issue,” she continued, “is to inform the patient with NF1 that they know how to recognise PN and its complications, and who they should go to for proper advice.” As such, Oostenbrink emphasised the importance of educating patients and caregivers that they need to say: “I have NF1,” so an HCP can then consider that the manifestation requires a specific approach.
PLEXIFORM NEUROFIBROMAS IN ADULTS
Diagnosis and treatment of PNs predominantly focuses on children, but there is growing awareness of the need for more monitoring, treatment, and care for adults. While young adults may especially want to feel independent of the need for monitoring, Oostenbrink warned that “this is the age that the manifestations can become higher risk if they are not monitored.”
She also discussed how there are patients who say: “I was told that if I didn’t have problems at puberty, there won’t be a problem later on.” She recounted that there are many examples where this is not true. For adults, there is a higher risk for malignancy in PNs,2 so it is important to educate patients on the need for follow-up when they are older, to monitor for complications and changes. It is also important to monitor adults with NF1, and associated PNs, to make sure complications do not arise, and to inform them about potential new treatments.
RESOURCES FOR PATIENTS AND HEALTHCARE PROFESSIONALS IN EUROPE
One valuable resource for patient education and support is dedicated NF1 organisations, to whom patients should be referred. As well as individual country organisations, Oostenbrink noted that there is also NF Patients United, which interconnects the patient organisations at a European Level.10 For HCPs, there is also the European Reference Networks (ERN) for Rare and Low Prevalence Complex Diseases, where NF1 is identified as a rare genetic tumour risk syndrome.11 This, Oostenbrink highlighted, also has patient representative and advocacy groups, and general information for patients. Additionally, she explained: “Some patient organisations have developed local materials to inform patients in their own language, which are now being copied, to make them more general for Europe.”
The ERN basic concept, explained Oostenbrink, is to make sure that in every country there is at least one access point to share care. While this is a valuable initiative, as not all centres in one country need to be included, it does not connect all HCPs with NF1 experience. To include such HCPs, Oostenbrink discussed how there is now the Children’s Tumor Foundation (CTF) Europe,12 which includes a clinical care advisory board, and a clinical care network. They aim to open the network to all centres with experience of NF1, not just expertise centres.
Both the CTF Europe13 and ERN GENTURIS have developed masterclasses to train and educate HCPs caring for patients with NF1, to which Oostenbrink contributes as an expert. There is also a biannual European NF meeting that connects HCPs with the clinical expertise of NF specialists, and researchers.
CONCLUSION
PNs may be a complex manifestation of NF1 to handle, and management requires a multidisciplinary approach, directed by a specialist centre and an MDT. Support also needs to be given to help patients cope with the medical and psychosocial burden of the disease, as this can be very high.
Though rare, most GPs should expect to see at least one patient with NF1. As such, they should be aware of how to recognise this condition, refer their patient(s) to a specialist centre, and potentially be involved with long-term monitoring.
European-wide HCP and patient organisations are connecting those less experienced in PNs with experts. These networks aim to enhance the recognition and care of people with PNs, and to discuss emerging therapies for those with inoperable PNs, such as mitogen-activated protein kinase inhibitors, which are currently being approved and may soon be widely available in clinical practice.7
In a follow-on article, Amedeo Azizi, Division of Neonatology, Pediatric Intensive Care and Neuropediatrics, Department of Pediatrics and Adolescent Medicine, Medical University of Vienna, Austria, discusses treatment options for PNs, and how these may evolve in the future.






