
Abstract
Primary cardiac tumours are exceedingly unusual and aggressive; they often develop in younger patients and present with advanced disease. The rarity and heterogeneity of primary cardiac tumours challenge the standardisation of therapeutic guidelines. Undifferentiated primary cardiac spindle cell sarcomas, a distinct subset of primary cardiac sarcomas, are especially unique with <20 cases reported worldwide, the majority of which are of left atrial origin. This article presents a review of the aetiology, pathophysiology, and therapy of undifferentiated primary cardiac spindle cell sarcomas. In conjunction, the authors present a unique case of a woman with hereditary nonpolyposis colorectal cancer (Lynch syndrome) who presented with a primary cardiac spindle cell sarcoma of left ventricular origin; this is the first case of this type and location of cardiac tumour reported in a patient with Lynch syndrome.
CARDIAC TUMOURS
Cardiac tumours are rare, with their incidence estimated between 0.0017–0.2800%,1 and the vast majority are metastatic lesions. Following benign myxomas, malignant primary sarcomas of the heart are the second most common cardiac tumour.1 Characteristics of malignant cardiac masses include broad-based growth, invasion of surrounding tissue, involvement of more than a single chamber, poor definition of borders, tissue inhomogeneity, large size (>5 cm), and the presence of pericardial or pleural effusion.2,3 Regardless of benign or malignant status, cardiac tumours are usually asymptomatic until they grow large enough to disrupt valvular function, obstruct a cardiac chamber,4 or invade the myocardium and conductive tissue to precipitate arrhythmias.5 Characteristics of cardiac tumours associated with increased survival include left-sided location, the absence of necroses or metastases, and low mitotic count. Notably, age, sex, differentiation, and histology have no effect on prognosis.6
The growing incidence of primary cardiac tumours over the past few decades is, in part, a reflection of advancements in cardiac imaging, such as echocardiography, MRI, and multidetector CT.7 Echocardiography can help to distinguish between different types of cardiac masses, including thrombi, vegetations, hypertrophy of the interatrial septum, and pericardial cysts.8 MRI and CT imaging can reveal invasive extracardiac extension of the sarcoma to help determine resectability and differentiate between benign and malignant lesions.9 Precise diagnosis is established by biopsy, although very few patients have a histological diagnosis prior to surgery.4 Despite a lack of standard treatment guidelines, most physicians advocate for early and total surgical tumour excision followed by adjuvant chemotherapy or chemoradiation.10
PRIMARY CARDIAC SARCOMA
Primary cardiac sarcomas are malignant tumours that originate from mesenchymal cells and are confined to the heart.11 They comprise 95% of all malignant primary cardiac tumours and are extremely rare and lethal.12,13 Some hypothesise that the poor prognosis associated with primary cardiac sarcoma is a function of both local invasion and metastases at presentation.14 A retrospective analysis of cardiac sarcomas reported a 0.0017% prevalence of cardiac sarcomas at autopsy.15 Most cardiac sarcomas carry a provisional diagnosis of a benign myxoma until intra-operative observation of invasive neoplasm prompts the suspicion of sarcoma.16
Most of the gene alterations associated with sarcomas are chromosomal translocations.17 Histopathologic analysis allows for directed use of immunohistochemistry and molecular techniques that are instrumental in establishment of diagnosis.18 Cardiac sarcomas typically have genetic profiles with recurrent alterations in MDM2, PDGFRA, or EGFR, which have potential as future therapeutic targets.19 Immunohistochemistry can aid in the classification of cardiac sarcomas and distinguish benign from malignant lesions.14 However, immunochemistry has a limited value in the setting of evaluation of a cardiac sarcoma due to a frequent lack of morphologically recognisable differentiation, as well as a lack of tissue-specific antigens.11
Left-sided cardiac tumours are less infiltrative than right-sided tumours and have a superior overall survival.20 Primary left-heart sarcomas most frequently originate in the left atrium.21 Systemic metastases due to cardiac sarcoma are detected in 80% of cases at the time of presentation.1 Although left-sided cardiac tumours are slower to metastasise, their tendency to grow into the left atrium and obstruct flow often leads to heart failure requiring urgent surgery for alleviation of symptoms.8 While loco-regional progression is the most common cause of death in patients with cardiac sarcoma, tumour grade is more predictive of the development of metastases.22
Due to the rarity of primary cardiac sarcoma, there are no randomised controlled trials or guidelines to determine optimal treatment. Therapy should consider age, performance status, prognosis, clinical symptoms, and tumour characteristics (histology, size, location, and extent of systemic disease), as well as previous thoracic radiotherapy.23 The majority of primary cardiac sarcomas are high-grade24 and treatment decisions are primarily guided by anatomic location rather than by tissue type.25 Poor prognostic factors include tumours with high mitotic index, evidence of tumour necrosis, or invasion of myocardial tissue.26 In the absence of metastases at diagnosis, surgical resection of a primary cardiac sarcoma is considered the gold standard therapy11 and is a major determinant of survival.8 While cardiac function can be preserved in up to 30% of right ventricular surgical resection, left ventricular resections are higher risk.27 Patients with extensive cardiac metastases, significant intracavitary disease, or uncontrolled disease are poor candidates for surgical resection.28 Less than half of patients with cardiac sarcoma are able to undergo a complete tumour resection.29 Obstacles to complete surgical resection include deep invasion of cardiac tissues or tumour location in a site that necessitates intricate reconstruction.11 As Putnam et al.30 concluded, “though operative mortality and morbidity may be high, complete resection will yield twice the long-term survival as that of unresectable patients with significant symptom-free survival.”
Patients with cardiac sarcoma who receive combination therapy, including surgery, radiation, and chemotherapy, demonstrate a 22.4-month average additional survival benefit in comparison with those treated with surgery, radiation, or chemotherapy alone.31 A retrospective analysis of surgically resected cardiac sarcomas found that low-grade tumour histology and survival following initial resection were associated with improved prognosis.32 While surgical resection is the preferred therapy for cardiac sarcomas, there is a mortality risk of 8.3%.30 Systemic therapy is frequently used in patients with incomplete surgical resections, recurrent tumours, or in the setting of an especially aggressive neoplasm.14 Some advocate that chemotherapy has a role even following a complete surgical resection due to the high likelihood of missed microscopic disease.25 If complete resection is unfeasible, cardiac transplantation may be considered in the absence of metastases. If the tumour is unable to be resected in the setting of metastases, palliative chemotherapy is recommended.33 Some patients with cardiac sarcoma receive chemotherapy including adriamycin and ifosfamide, or gemcitabine and docetaxel.8
While cardiac sarcomas are typically resistant to radiotherapy, sometimes radiotherapy is administered in large high-grade tumours or those with positive or borderline margins following surgical resection.12 Although radiotherapy has been associated with decreases in local recurrence rates, there is a risk of myocardial damage, including cardiomyopathy and pericarditis, along with minimal or no influence on survival.14 In the absence of metastases, both orthotopic cardiac transplantation (receipt of a donor’s heart) and cardiac autotransplantation (cardiac excision with ex vivo tumour resection and re-implantation) are potential therapies in primary cardiac sarcoma.20,25 Some providers are hesitant to recommend cardiac transplantation given the potential for immunosuppressant therapy to potentiate metastases.29 In addition, immunosuppression has the potential to induce new neoplastic growth, thereby catalysing the development of another primary malignancy.14 However, historical data demonstrated that orthotopic heart transplantation has a long-term survival benefit without recurrence, despite use of immunosuppressants.34
UNDIFFERENTIATED SPINDLE CELL SARCOMA
Intimal cell sarcoma is a subtype of undifferentiated spindle cell sarcoma, a large and diverse group of tumours composed of elongated cells that arise from the inner layer of the arterial wall. This class of sarcoma is particularly heterogeneous, with variance among their structural composition of nuclei shape, volume of cytoplasm, stromal fibrosis, and myxoid accumulation.35 Spindle cell sarcomas are usually immunoreactive to vimentin, osteopontin, and MDM212,36 with variable positivity for alpha smooth muscle actin, CD117, CD68, p53, and Bcl-2.16 Complex karyotypes of the MDM2 gene encompassing the 12q13-14 region are usually present in spindle cell sarcoma.37 Spindle cell sarcoma lacks a specific and universal histological grading scheme, complicating its diagnosis.38
According to an update of the World Health Organization (WHO) 2013 guidelines, spindle cell sarcomas fall into the undifferentiated/unclassified subcategory of sarcomas and account for up to 20% of sarcomas.39,40 Undifferentiated sarcomas are tumours in which all recognisable tissue differentiation has been excluded;40 they are further classified by principal type of morphology including round cell, spindle cell, pleomorphic, and epithelioid.40 Undifferentiated sarcomas are particularly aggressive and high-grade. Remarkably, 80% of patients with spindle cell sarcoma have metastases at the time of diagnosis.16 Unfortunately, due to its rarity, the management of spindle cell sarcoma is driven mostly by anecdotal data with a lack of evidence-based guidelines.16 As a result of the rare incidence of spindle cell sarcoma, distinct clinical characteristics, outcomes, organ involvement, and specific prognostic factors remain enigmas.38 Spindle cell sarcomas typically form polypoid masses that attach to the inner surface of a blood vessel in a manner comparable to a thrombus.13 Varying expression of cell-cycle markers, specifically a decrease of p27Kip1 expression and an increase of cyclin E expression, may account for the aggressive and invasive nature of spindle cell sarcomas.
PRIMARY CARDIAC SPINDLE CELL SARCOMA
Primary cardiac spindle cell sarcoma is the rarest form of primary cardiac tumour, with very few publicised reports and non-existent treatment guidelines.41 According to the authors’ knowledge, only 15 cases (Figure 1) of primary cardiac spindle cell sarcoma have been reported worldwide. They are distributed between 12 left atrial tumours,37,42-52 1 left ventricle tumour,53 1 right ventricle tumour,16 and 1 pericardial tumour.54
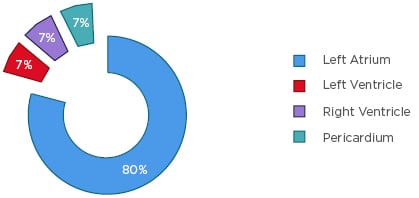
Figure 1: Anatomic distribution of primary cardiac undifferentiated spindle cell sarcoma.
The mesenchymal origin of cardiac spindle cell sarcomas explains their predilection to affect large cardiopulmonary blood vessels.16 Spindle cell sarcomas are typically positive for complex karyotypes encompassing the 12q13-14 region (MDM2 gene).37 Immunochemistry stains can detect MDM2 in cardiac sarcomas that are poorly differentiated and may be confirmed by further studies, including fluorescence in situ hybridisation (FISH), quantitative PCR, or array comparative genomic hybridisation analysis.19 The prognosis of cardiac primary spindle cell sarcomas is poor with a mean survival of 3 months to 1 year.16 Initial symptoms include blood flow obstruction, embolism, and regional neoplastic infiltration.16 Presenting symptoms of left atrial primary cardiac spindle cell sarcoma include high fever, mass effect on mitral valve leading to mitral stenosis, and severe congestive heart failure.55
Treatment strategies of cardiac spindle cell sarcomas vary and may involve surgery, chemotherapy, and radiation. Regardless of differing surgical techniques, patients with primary cardiac spindle cell sarcoma who underwent surgical resection had more favourable disease-free survival and overall survival in comparison with those who did not receive surgery,38 which was true regardless of the quality of resection or presence of metastases.56 Even following a complete surgical resection, patients with primary cardiac spindle cell sarcoma most commonly died from macroscopic local tumour.43
LYNCH SYNDROME
Lynch syndrome, also referred to as to as hereditary nonpolyposis colorectal cancer, is an autosomal dominant disorder resulting from germline mutations in the mismatch repair genes (MMR) including MLH1,57 MSH2,58 and MSH6.59,60 Patients with MLH1 and MSH2 mutations have a higher risk of malignancy (specifically colorectal cancer) than those with MSH6 mutations.61 Interestingly, the rate of ovarian cancer increases markedly at age 40 years for patients with mutations in MLH1 and MSH2.61 Generally, somatic mutation of an MMR gene in a patient with Lynch syndrome results in an increased number of DNA replication errors; these errors are a reflection of the degree of microsatellite instability: i.e., differing lengths of repetitive DNA sequences in tumour-extracted DNA.62 Defects in mismatch repair genes predispose patients to develop different types of cancer with a guiding symptom of development of usually right-sided colorectal cancer at a young age.63 Despite the frequency of right-sided colorectal cancer in patients with Lynch syndrome, other neoplasms have also been reported in higher frequencies in patients with Lynch syndrome and include tumours of the stomach, ovaries, pancreas, biliary tract, brain, sebaceous gland adenomas, and keratoacanthomas.64
Sarcoma in Lynch Syndrome
Soft tissue sarcomas are neither Lynch syndrome-defining malignancies nor neoplasms that are commonly associated with Lynch syndrome.65 Diagnosis of a Lynch syndrome-related neoplasm is achieved through microsatellite instability testing of the sarcoma by immunohistochemical staining of MMR proteins.65 Traditionally, the development of sarcomas is due to alteration of tumour suppressor genes rather than alteration of oncogenes.66 However, sarcomas may emerge in the setting of Lynch syndrome.67 While a few sarcomas, including leiomyosarcoma and undifferentiated pleomorphic sarcoma, have been associated with Lynch syndrome, liposarcoma is the most frequent.67-72
A prospective study of 658 adults with soft tissue sarcoma revealed that 2.8% of those patients had a genetic syndrome, predominantly Recklinghausen neurofibromatosis and bilateral retinoblastoma.73 While reports of rhabdomyosarcoma67 and gastric sarcoma74 have been identified in patients with Lynch syndrome, no reported cases of cardiac sarcoma or spindle cell sarcoma have been previously reported in patients with either constitutional mismatch-repair deficiency or Lynch syndrome.70 Documented cases of soft tissue sarcomas in patients with Lynch syndrome families68,75 may be associated with mutations in MSH2, MSH6, and MLH1.62,68,69 An analysis of 11 patients with Lynch syndrome and soft tissue sarcomas revealed a germline MSH2 mutation in over half of the cases. Despite a small number of cases, the authors hypothesise that the risk of soft tissue sarcomas is higher in MSH2-deficient patients and families.71 Notably, patients with a personal or familial history of Lynch syndrome who develop sarcomas demonstrate a predominance of MSH2 mutations.70 While some researchers hypothesise the association between soft tissue sarcomas and MMR gene deficiency, particularly hMSH2,71 others question the influence of microsatellite instability and mismatch repair mutations as features of sarcomas.67 Soft tissue sarcomas in patients with Lynch syndrome have previously been considered coincidental.69 Further investigation of patients with Lynch syndrome and sarcoma is needed, with precise analysis of mutated MMR genes including MLH1, MSH2, MSH6, PMS2, as well as the p53 and retinoblastoma signalling pathways.68
CASE REPORT
A 55-year-old woman with a past medical history of Lynch syndrome and ovarian clear cell carcinoma after total abdominal hysterectomy and bilateral salpingo-oophorectomy and six cycles of carboplatin and paclitaxel presented to a local hospital with acute dyspnoea. Family history was remarkable, with a father, paternal uncle, and paternal aunt with colon cancer, as well as a paternal aunt with ovarian cancer. The patient received screening colonoscopies every 3 years that were unremarkable, most recently in November 2017. The patient worked as an administrator and denied tobacco, alcohol, or drug use.
Three months prior to presentation, the patient reported a few-week history of viral respiratory illness that precipitated an acute episode of chest pain, dyspnoea, and near-syncope. The patient was taken to a local hospital where she was found to be in cardiogenic shock (blood pressure 61/43 mmHg) requiring vasopressors. Laboratory testing showed leukocytosis of 18.4 thousand/uL, haemoglobin 13.8 g/dL, alanine aminotransferase 115 U/L, aspartate aminotransferase 124 U/L, and lactic acid 4.4 mmol/L, with the remaining laboratory results within normal limits. Troponin <0.02 ng/mL, brain natriuretic peptide 18 pg/mL, D-dimer 193 ng/mL, and lipase 141 unit/L were all negative. Physical exam was remarkable only for jugular venous distension. CT scan of the chest demonstrated a large pericardial effusion and an echocardiogram revealed a massive circumferential pericardial effusion with early diastolic collapse of the right atrium consistent with early tamponade. A subxiphoid pericardial window placed for cardiac tamponade, fluid cytology was negative, and the patient was discharged with a suspected viral pericarditis.
One month later, screening breast ultrasound and mammogram revealed an irregular hypoechoic mass 9x9x9 mm with two adjacent irregular hypoechoic nodules overlying the right pectoralis muscle. Previous mammograms, most recently 6 years prior, were without any features to suggest malignancy. Over the next few weeks, the patient developed recurrent acute dyspnoea, along with palpitations and chest pain. A physical exam showed a pericardial rub and a right breast mass in the upper-outer quadrant with associated palpable ipsilateral lymphadenopathy. CT showed a moderate left pleural effusion and a large pericardial effusion with mass effect on the main pulmonary artery, left ventricle, and left pulmonary veins (Figure 2). Transthoracic echocardiogram (TTE) revealed a small and distorted left ventricular cavity, compression of left ventricular wall by a large 10–12 cm loculated semiliquid pericardial mass/effusion, and compression of the main pulmonary artery with peak gradient of 25–30 mmHg. Left anterior mini-thoracotomy revealed an avascular multilobulated mass with left ventricular myocardial invasion and associated tamponade. Additionally, 800 mL of pericardial fluid and 2 L of haemorrhagic left pleural fluid were removed. A biopsy of the left ventricular mass revealed myxoid spindle cell sarcoma of uncertain histogenesis and biopsy of intrapericardial tissue demonstrated benign pericardium with patchy chronic inflammation and fibrinous exudate (Figure 3). Immunostains for calretinin highlighted reactive mesothelial cells in the pericardial tissue and immunostains were negative for Ber-Ep4, MOC-31, MDM2, CDK4, CK, B100, SOX10, CD34, and desmin. NTRK1 immunostain was positive; however, FISH studies did not reveal any genomic abnormality in NTRK1 or NTRK3. Simultaneous biopsy from the right breast revealed poorly differentiated invasive ductal carcinoma that was oestrogen and progesterone receptor negative with an equivocal human epidermal growth factor receptor 2 score (2+) and a Ki67 of 50%. Immunostains showed tumour cells positive for GATA-3, E-cadherin, CK5, and p63 (patchy). Biopsy of axillary lymph nodes was consistent with metastatic carcinoma from tumour in concurrent breast.
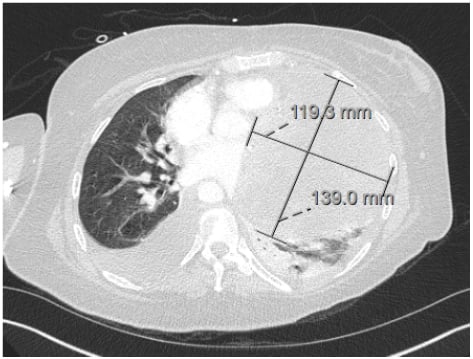
Figure 2: CT showing moderate left pleural effusion and a large pericardial effusion with mass effect on the main pulmonary artery, left ventricle, and left pulmonary veins.
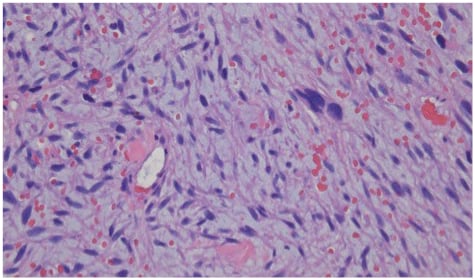
Figure 3: Biopsy of intrapericardial tissue demonstrating benign pericardium with patchy chronic inflammation and fibrinous exudate.
The patient was diagnosed with cardiac spindle cell sarcoma (based on sections cT3, cN0, cM0 of the Soft Tissue Sarcoma of the Abdomen and Thoracic Visceral Organs staging form)76 and was referred for chemotherapy with gemcitabine and docetaxel. Treatment of the cardiac sarcoma was prioritised over the 1.3 cm malignant breast tumour given progressive and debilitating cardiopulmonary symptoms, although the regimen of gemcitabine and docetaxel was selected to treat both. One week following induction chemotherapy, the patient’s dyspnoea progressed from exertional-only to present at rest and she presented to the authors’ emergency department for further evaluation. Vital signs on arrival were 114/71 mmHg, heart rate of 70 beats per minute, respiratory rate of 20 breaths per minute, temperature 98 °F, and oxygen saturation was 97%. Laboratory tests were notable for lactic acid 1.5 (mmol/L), troponin <0.011 (ng/mL), and brain natriuretic peptide 2,100 (pg/mL). Chest X-ray with markedly increased large left pleural effusion, increased moderate right pleural effusion, and new superimposed reticulonodular opacity in the right upper lung could. TTE revealed a left ventricular ejection fraction of 60–65%, redemonstration of a large mass extrinsic to the heart in the posterolateral region of the left ventricle, mildly elevated pulmonary artery systolic pressure (31 mmHg), and moderate left ventricular hypertrophy.
Thoracentesis of the left chest yielded 700 cc reddish fluid with reactive mesothelial cells and lymphocytes, without evidence of malignancy. Repeat TTE showed a small left ventricle, left ventricular ejection fraction 60–65%, a small pericardial effusion, an underfilled right ventricle, moderate tricuspid regurgitation, and moderately elevated pulmonary artery systolic pressure (41 mmHg). Given the rapid development and progression of the tumour with severe functional limitations in the setting of the low likelihood of complete surgical resection, the family pursued a palliative approach. The patient died days later, 3 months following initial symptoms and 4 weeks following diagnosis.
CASE DISCUSSION
While there has been only one case, to the authors’ knowledge, of a pulmonary artery spindle cell sarcoma in a patient with Lynch syndrome,77 there are no documented cases of either primary cardiac sarcoma or primary intracardiac spindle cell sarcoma in patients with Lynch syndrome. Moreover, this case represents the first MDM-negative primary cardiac sarcoma ever reported. In addition to the negative MDM immunohistopathologic analysis, this tumour was also negative for vimentin and desmin, which, along with MDM, are immunoreactive in spindle cell sarcoma and serve as principle diagnostic markers.12,36,45
To the authors’ knowledge, there are only 15 reported cases of primary intracardiac spindle cell sarcoma, 80% of which are tumours of the left atrium. The left ventricular origin of this primary cardiac spindle cell sarcoma differentiates it from most cardiac sarcomas and other primary cardiac spindle cell sarcomas as the second such published case.53 Some elements of this case presentation were typical of primary cardiac tumours, including haemodynamic dysfunction with induction of cardiac tamponade and initial presentation with New York Heart Association (NYHA) Class IV symptomatology. However, the absence of metastases at diagnosis is atypical for a left-sided cardiac sarcoma. With >80% of patients with primary cardiac spindle cell sarcoma presenting with metastases at diagnosis,16 this case defies typical pattern and progression of disease. Left-sided cardiac tumours are typically less invasive and associated with higher rates of overall survival. This cardiac tumour was extremely aggressive with a 3-month survival time following initial symptoms, with sequential echocardiograms illustrating the 4-week development of a mass with compression of both the lateral left ventricular wall and main pulmonary artery. The dimensions of this cardiac mass were particularly large, with a diameter of 12 cm, more than twice the average 5 cm size of a cardiac tumour.78 Divergent from the tendency of left-sided cardiac tumours to have less invasive growth and improved survival, this case demonstrated rapid and extensive neoplastic infiltration with corresponding severely diminished survival. The simultaneous recognition of two separate primary malignancies not commonly associated with Lynch syndrome, breast carcinoma, and cardiac spindle cell sarcoma, further differentiate this case. The large, invasive mass induced cardiogenic shock and was so intricately intertwined with the left ventricular myocardium that it created an unfeasible surgical resection. Careful selection of chemotherapy agents to treat both the breast cancer and the cardiac sarcoma were considered and the combination of gemcitabine and docetaxel was selected.
CONCLUSION AND FUTURE DIRECTIONS
The authors report the first case of MDM negative primary cardiac sarcoma and the first case of primary intra-cardiac spindle cell sarcoma in a patient with Lynch syndrome. To the authors’ knowledge, this is the second case of left ventricular primary cardiac spindle cell sarcoma published to date. The rapidly progressive lethality of spindle cell sarcoma in conjunction with the haemodynamic consequences of cardiac tumours form a hazardous combination with severe implications on cardiac function and patient symptomatology. While sarcoma is not a common cancer in patients with Lynch syndrome, those patients with a family history or personal diagnosis of an MMR deficiency should undergo routine surveillance for Lynch syndrome-related malignancies, including regular colonoscopies. While the rarity of primary cardiac sarcomas precludes the establishment of specific treatment algorithms, further research is needed to optimise therapies and the presence of sarcomas in Lynch syndrome may contain additional links not currently identified. Future studies should emphasise genetic analysis to identify common genes to predict risk of neoplastic development, as well as subsequent gene-specific treatment protocols to treat malignancies.




