
Abstract
Worldwide, head and neck squamous cell carcinoma (HNSCC) is responsible for >550,000 diagnoses and 380,000 deaths annually. It originates in the upper aerodigestive tract and has a multifactorial origin involving both genetic and lifestyle risk factors. The clinical management of HNSCC involves surgery, radiotherapy, and chemotherapy. Several studies point to the role of genetic variations in predicting drug efficacy and toxicity. Cancer pharmacogenomics has fast emerged as a new and promising field for the early identification of genetic markers that can predict drug response or toxicity, with the number of studies of genetic polymorphisms as prognostic factors of HNSCC treatment outcomes growing. The number of studies evaluating the association of candidate polymorphisms in drug-metabolising Phase I and II enzymes with treatment outcome far exceed the studies involving other candidate genes, such as those involved in drug metabolism, DNA repair, and cell cycle regulation. This review focusses on the relevance of genetic variations in genes, where the corresponding gene products play an important role in drug metabolism (TPMT, DPD), DNA repair (X-ray repair cross complementing 1), cell cycle (tumour protein P53), and carcinogenesis (matrix metalloproteinase 3 and 7), thereby contributing to the treatment outcome for HNSCC. This could greatly help clinicians in identifying genetic markers useful for the selection of optimal drugs, dose, and treatment duration on an individual basis, resulting in improved drug efficacy and decreased toxicity. However, further studies are needed in well characterised and larger HNSCC populations with proper validation of pharmacogenetic markers in experimental settings before application in clinical routine diagnostics.
INTRODUCTION
It has generally been observed amongst patients that a medication proven efficacious for the majority of patients often fails to work in some other patients. In those cases where it works, it may cause serious side effects, even death, in a small number of patients. It is now well established that a large variability of drug efficacy and adverse drug reactions in patients decides the clinical use, regulation, and market withdrawal of clinical drugs.1 Similar observations have also been made for cancer medication as significant heterogeneity in the efficacy and toxicity of chemotherapeutic agents has been found to exist across the human population.2
Recent studies from our laboratory have shown that treatment response in head and neck squamous cell carcinoma (HNSCC) patients treated with a combination of chemotherapy and radiotherapy regimen of combination drugs, such as cisplatin and 5-fluorouracil (5-FU), was poor in patients with variant genotypes of CYP2C9, CYP2C19, and CYP2D6.3,4 Furthermore, an association with poor treatment response to chemotherapy was also observed in HNSCC cases for genotypes of CYP2A6, GSTM1, GSTT1, and GSTP1.5,6 Apart from the Phase I and Phase II drug metabolising enzymes, studies were also conducted to evaluate the role of genetic variants in other drug metabolising enzymes, such as thiopurine S-methyltransferase (TPMT) and dihydropyrimidine dehydrogenase (DPD), in determining the treatment outcome in HNSCC patients.7 This review will focus on some of the major genetic variations in TPMT, DPD, and some of the other key target genes, such as those involved in DNA repair (X-ray repair cross complementing 1 [XRCC1]), cell cycle (tumour protein P53 [TP53]), and carcinogenesis (matrix metalloproteinases, [MMP]), which might play an important role in determining the treatment outcome in HNSCC cases. The genomic structures of TPMT, DPD, and XRCC1 are shown in Figure 1, whilst Table 1 lists the single nucleotide variants associated with drug response.8
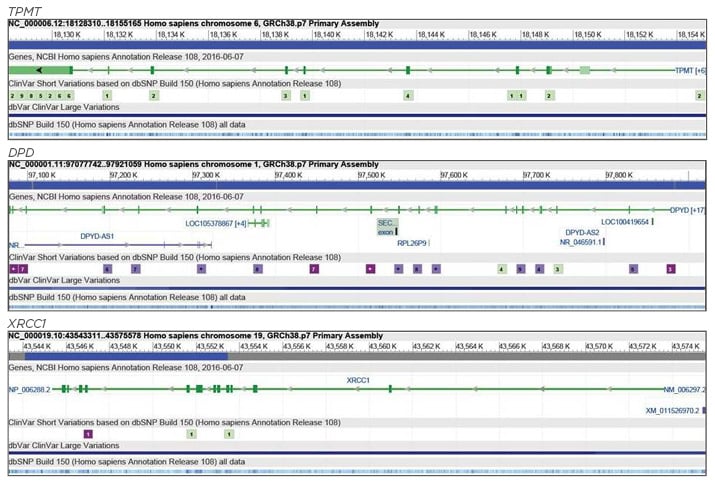
Figure 1: Genomic structures of TPMT, DPD, and XRCC1 genes.
DPD: dihydropyrimidine dehydrogenase; TPMT: thiopurine 5-methyltransferase; XRCC1: X-ray repair cross complementing 1. Adapted from National Center for Biotechnology Information (NCBI).8
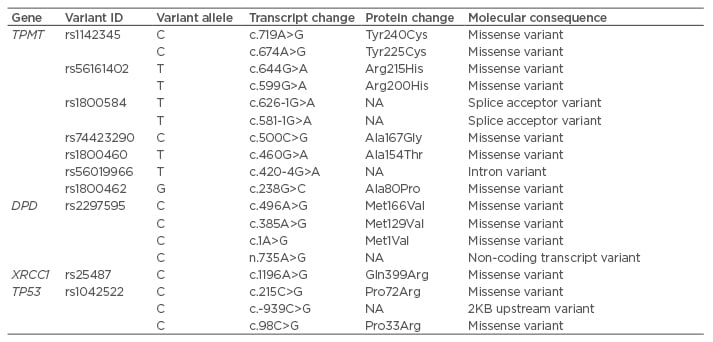
Table 1: Table showing the single nucleotide variants associated with drug response.
DPD: dihydropyrimidine dehydrogenase; NA: not applicable; TPMT: thiopurine 5-methyltransferase; TP53: tumour protein 53; XRCC1: X-ray repair cross complementing 1.
DRUG METABOLISM POLYMORPHISMS: THIOPURINE-S-METHYLTRANSFERASE AND DIHYDROPYRIMIDINE DEHYDROGENASE
TPMT and DPD are enzymes involved in the metabolism and disposition of drugs, such as 6-mercaptopurine, azathioprine, cisplatin, 5-FU, and 6-thioguanine, that are used for the treatment of different cancers, autoimmune disorders, dermatological conditions, and as an immunosuppressant in graft transplantations.9-12 TPMT functions by methylating mercaptopurine and thereby reducing its bioavailability for conversion into thioguanine nucleotides, which is the cytotoxic form of the drug. Approximately 10% of patients have an intermediate enzyme activity and have a greater incidence of thiopurine toxicity, while 0.3% are deficient of TPMT activity, having severe or fatal toxicity from mercaptopurine therapy.13
Genetic variants present in TPMT may alter the treatment response in cases receiving chemotherapeutic drugs. The four major variant alleles that lead to 80–95% intermediate and low enzyme activity of the TPMT gene are TPMT*2 (238G>C), *3A (460G>A and 719A>G), *3B (460G>A), and *3C (719A>G).14 TPMT*3A mutant allele contains two nucleotide transitions, 460G>A and 719A>G in the open reading frame, leading to the substitution of amino acids Ala154Thr and Tyr240Cys, respectively. Our study has shown that the treatment response after receiving cisplatin and 5-FU was modified in patients who carried variant genotypes of TPMT*3B and *3C.7 Since individuals with TPMT*2, *3A, *3B, and *3C variants are reported to be associated with lower or intermediate enzyme activity,15,16 the poor response rate in patients could probably be a result of decreased TPMT activity, which may lead to relatively higher intracellular concentration of cisplatin and therefore higher incidence of cisplatin-related toxicity. Liu et al.17 performed a genome-wide association study of primary erythrocyte TPMT activity in children with leukaemia (N=1,026) and found that TPMT was the only gene that reached genome-wide significance (top hit rs1142345 or 719A>G; p=8.6×10-61). Furthermore, TPMT activity in patients behaves as a monogenic trait, further corroborating the importance of TPMT genetic testing in clinical settings.
The DPD gene also has several reported polymorphisms, out of which consistent data are available for the DPD*2A allele, which is caused by a G>A transition at a GT splice donor site flanking exon 14 of the DPD gene (IVS14+1G>A).18 This allele is the most common and results in the elimination of a 55-amino-acid-long fluorouracil binding site in the DPD protein, leading to the skipping of exon 14 and the production of an inactive protein in 0.9% of the Caucasian population.19-21 Both anabolic and catabolic pathways are involved in the cytotoxic effects of 5-FU;9 thus the activity of DPD plays a role both in determining the efficacy of the therapy with 5-FU and its toxicity.22-24 Decreased DPD activity has been associated with >4-fold risk of severe or fatal toxicity from standard doses of 5-FU.25 DPD has another mutation at codon 534, leading to a 1601G>A nucleotide change.26 Our study has also shown that cases carrying the DPD genetic variant IVS14+1G>A also showed a poor treatment response.7 Other studies also reported that a high prevalence of IVS14+1G>A was associated with increased toxicity and poor treatment response in patients of invasive ductal carcinoma and Grade 3 and 4 toxicity in HNSCC patients.27,28 Amongst the reasons for the toxicity of 5-FU are its relatively narrow therapeutic index29,30 and low DPD activity. Lowered DPD activity leads to less effective inactivation of 5-FU, causing an accumulation of excessive levels of fluorodeoxyuridine monophosphate, causing gastrointestinal, haematopoietic, and neurological toxicities that are potentially fatal.21 Zhao et al.31 investigated the association between germline genetic polymorphisms in DPD and outcome of acute lymphoblastic leukaemia following the treatment with 5-FU plus oxaliplatin, and found that the common DPD variant c.85T>C (rs1801265, DPYD*9A) was significantly associated with complete response rate, event-free survival, and treatment-related toxicity.
DNA REPAIR GENE POLYMORPHISMS: X-RAY REPAIR CROSS-COMPLEMENTING GROUP 1
The DNA damage induced by anticancer agents is repaired by the DNA repair enzymes and, therefore, it can be hypothesised that polymorphisms in DNA repair genes may influence the outcome of treatment. Increased DNA damage may prove beneficial in treatment, as both chemotherapy and radiation rely on DNA damage as part of their mechanisms of tumour cell destruction though the same DNA damage that occurs within the tumour may also take place in normal tissues as a side effect of therapy. XRCC1 is a major DNA repair gene in the base excision repair pathway and is involved with radiation-related DNA repair. The base excision repair pathway repairs single strand breaks through protein-protein interactions, by interaction of XRCC1 with PARP-1, PNK, Polb, and Lig3a.32 XRCC1 interacts with PNK, leading to the stimulation of both the 5’-kinase and 3’-phospatase activities of PNK; the interaction with Lig3a increases the intracellular stability of the ligase.32 Several single nucleotide polymorphisms (SNP) in XRCC1 are reported, out of which the most extensively studied are Arg194Trp on exon 6 (dbSNP no. rs1799782), Arg280His on exon 9 (dbSNP no. rs25489), and Arg399Gln on exon 10 (dbSNP no. rs25487).33
A study conducted by Quintela-Fandino et al.34 tested the ability of XRCC1-Arg399Gln gene polymorphism to predict response to cisplatin in peripheral blood mononuclear cells of Stage-IV HNSCC patients. XRCC1 Gln/Gln was associated with 0% of cases with progressive cancer, 7.7% of cases with stable disease, 30.8% of cases with partial response, and 61.5% of cases with complete response. Duell et al.35 reported that the rate of sister chromatid exchange after exposure to ionising radiation in human lymphocytes was greater in individuals with the XRCC1-Gln399Gln genotype than in those with only wild-type genotype. Alsbeih et al.36 tested the association between XRCC1 G28152A Arg399Gln (rs25487) SNP and late reaction to radiotherapy in 60 nasopharyngeal cancer patients. Significant association was observed between XRCC1 G28152A genotype (p≤0.05) and a lower grade of fibrosis (odds ratio:0.30; 95% confidence interval:0.10–0.89; p=0.02), and therefore the wild-type was the risk allele. However, Mahimkar et al.37 did not observe a significant association between polymorphisms of XRCC1 (Arg194Trp, rs1799782 and Arg399Gln, rs25487) and clinical outcome in advanced oral cancer patients treated with postoperative radiotherapy. Zhai et al.38 investigated the association of XRCC1 Codon399 polymorphism with treatment efficacy and normal tissue toxicity in Chinese patients with locally advanced nasopharyngeal carcinoma after radiotherapy. It was observed that Codon399 Gln/Gln allele correlated with a higher medium-term tumour regression ratio after radiotherapy for primary nasopharyngeal neoplasm and metastatic lymph nodes (>80% versus 40–60%; p<0.01). Compared with the other two genotypes in this study, patients with XRCC1 Codon399 Gln/Gln allele were more likely to obtain complete remission of the tumour (100% versus 76% and 67%; p>0.05). However, no correlation between XRCC1 Codon399 polymorphisms and acute or late radiation-induced injury of normal tissues was observed. Ghazali et al.39 conducted a systematic review of the association of SNP with the occurrence of HNSCC radiotoxicity and evaluated the association of 11 polymorphisms in 8 genes with acute radiotoxicity, and 6 polymorphisms in 4 genes for late radiotoxicity. The risk of severe acute mucositis was associated with the G allele of XRCC1 (1196A>G) in patients treated with radiotherapy alone or chemotherapy.
CELL CYCLE AND MATRIX METALLOPROTEINASES POLYMORPHISMS:TUMOUR PROTEIN P53 (TP53) AND MMP3, MMP7
The p53 protein, rightly known as the ‘guardian of the genome’, or the ‘cellular gatekeeper’, acts by transmitting a variety of stress-inducing signals to different antiproliferative cellular responses. Some major events such as DNA damage, oncogene activation, or hypoxia can lead to p53 activation, thereby regulating processes such as apoptosis, cell-cycle arrest, senescence, or modulation of autophagy.40 Somatic mutations of the tumour suppressor gene p53 occur in around 50% of all human cancers; thus, p53 is a major player in carcinogenesis.41 In addition to its role in carcinogenesis, p53 also plays a major role in deciding the clinical outcome of cancer treatment, because p53 mutations render tumour cells resistant to chemotherapy.42,43 The two p53 homologues, namely p63 and p73, are also activated by cytostatic drugs.44 p73 also functions in transduction of specific types of DNA damage, as activation of the p73/c-abl pathway by some chemotherapeutic agents causes apoptosis via target genes, such as AIP1.45,46 p73 exists as several splice variants at both N and C termini, with N-terminal variants lacking the transactivation domain and thereby acting as dominant-negative inhibitors of both p53 and p73.47 Out of the polymorphisms encoding either arginine (72R) or proline (72P) at codon 72 of p53, 72R is detected more commonly than 72P in squamous cell carcinomas.48,49 There is an over-representation of 72R mutants in carcinomas, which can be explained by the fact that inhibition of p73 is influenced by the polymorphism at codon 72 of p53.
Bergamaschi et al.50 reported that the p53-p73 interaction and its regulation by p53 polymorphism is an important determinant of response and survival in head and neck cancer. Patients with specific p53 mutations in the 72R form are least likely to gain complete response to chemotherapy. Upon treatment with cisplatin-based combination therapy, HNSCC cases with the 72R SNP has a worse response than those with the 72P SNP.50 Sullivan et al.51 reported that the response of human H1299 p53 -/- cells to anticancer agents is strongly influenced in vitro by a SNP in wild-type p53. The clinical relevance of these observations was presented by the evidence that the superior activity of the 72R wild-type protein is reflected in vivo by significantly higher response rates and survival in patients receiving chemo-radiotherapy for advanced head and neck cancer, who express the 72R wild-type variant. Seventy patients with inoperable advanced HNSCC who received cisplatin-based chemo-radiotherapy were involved in the study. All the patients presented with locally advanced, unresectable, tumour, node, and metastasis Stage III/IV HNSCC. In the 43 patients who had a wild-type p53 allele and had an evaluable response to treatment, complete response appeared significantly linked with p53 status (wild-type 72R p53 allele, wild-type 72P p53 allele, or both). Wild-type 72R allele produced the highest complete response rate (27 out of 28 patients [96%]). Similarly, overall survival and progression-free survival were significantly longer in patients with wild-type p53 allele (either 72R or 72P) than in those lacking a wild-type allele. Significant differences for overall survival and progression free survival were observed between cases retaining a wild-type 72R allele, cases retaining a wild-type 72P allele, and cases retaining both wild-type 72R and 72P alleles with the best prognosis in cases retaining a wild-type 72R allele. These clinical results corroborate the in vitro data wherein the expression of 72P wild-type p53 resulted in G1 cell cycle arrest with minor induction of apoptosis after challenge with drug doses that cause extensive apoptosis in 72R-expressing variants.50
MMP are a family of proteins involved in extracellular matrix degradation, cell proliferation, and angiogenesis, the processes that are involved in carcinogenesis.52 Fas ligand (FasL) is a member of the tumour necrosis factor superfamily that induces apoptosis in susceptible cells upon cross-linking of its own receptor, FasL (Apo-1/CD95), which contributes to immune homeostasis and cell-mediated cytotoxicity. FasL is upregulated in cells treated with genotoxic agents such as cisplatin and 5-FU, and thus it can be expected that the downregulation of FasL at the surface of HNSCC tumour cells decreases the activity of anticancer drugs through modulation of the apoptotic pathway. Studies suggest that MMP7 and MMP3 could be implicated in the shedding of FasL from the cell surface, resulting in the generation of soluble FasL with less effective activity for triggering apoptosis by crosslinking with Fas.53-55 This was experimentally shown by the work of Mitsiades et al.56 in Ewing sarcoma and colon carcinoma cells. Blockade of MMP7 activity by MMP inhibitor resulted in sensitisation to treatment with doxorubicin, providing evidence for the role of MMP and FasL in chemotherapy resistance.
Varying expression patterns of MMP could be due to the existence of functional genetic polymorphisms in MMP promoter regions. The two polymorphisms that result in increased promoter activity of MMP7 are one at position -181A>G and the other at position -153C>T. For MMP3, two alleles have been defined:the deletion (5A) or the insertion (6A) of an adenine at position -1612 (-1612insA), with the 6A allele being associated with low transcription levels.57 Amongst other MMP3 polymorphisms, six are in linkage disequilibrium with -1612insA and one (-A709G) has not been assigned functional importance.58 Blons et al.59 reported a correlation between MMP3 promoter polymorphism and response to chemotherapy in HNSCC. It was shown, for the first time, that patients carrying the low transcription level allele 6A had a better response to 5-FU-cisplatin combination therapy than patients carrying the 5A allele. Furthermore, multivariate analysis, including TP53 mutational status, showed that MMP3 was an independent factor in response to treatment. The results were in accordance to a study which showed that the induction of FasL after DNA damage is p53 independent.60 However, MMP7 promoter polymorphisms did not influence response to treatment.59
The MMP have been postulated to interact with the Fas/FasL pathways. Poulaki et al.61 extensively investigated the involvement of the FasL/Fas pathway in doxorubicin induced apoptosis in a model of Ewing’s sarcoma. Clones of the Fas-sensitive, doxorubicin-sensitive Ewing’s sarcoma cell line SK-N-MC, which were either Fas-resistant or FasL-deficient, were significantly resistant to Dox. It was also observed that cleavage of FasL by MMP7 (matrilysin) protected the parental SK-N-MC cells from doxorubicin, whereas inhibition of MMP7 activity increased their sensitivity. Gastman et al.62 provided a unique observation for the Fas/FasL pathway. The study showed that the pathway can be potentially immunosuppressive and may be involved in the escape of human carcinoma cells from immune destruction. The study found that the expression of FasL on HNSCC cell lines was upregulated by pretreatment of tumour cells with the metalloproteinase inhibitor, BB-94. When PCI-cell lines pretreated with BB-94 were co-incubated with Jurkat cells, they induced DNA fragmentation in a greater proportion of lymphocytes than did tumour cells not pretreated with the inhibitor.
In conclusion, genetic variations in genes encoding drug metabolising enzymes have explained a great deal of interindividual variation in response and toxicity of anticancer drugs. However, drug metabolism is just one aspect of the interaction of drugs and genes and more efforts should also be made to uncover the role of genetic variations in other important target genes, such as drug transporters, drug receptors, DNA repair, cell cycle, and other pharmacodynamic genes. Reduction of the toxicogenetic and toxicogenomic side effects has been one of the major goals in the search for new anticancer drugs and therapy protocols. Genome-wide linkage analyses may be a more systematic approach to discover genomic regions likely to carry genes, which determine chemotherapy cytotoxicity rather than the candidate gene-based approaches. Efforts should be made to implement SNP genotyping into clinical diagnostics, therefore allowing practitioners to make more accurate decisions for treatment and risk assessment. This will help in clinical decision making regarding treatment strategies, with the goal of avoiding adverse drug reactions while achieving the best drug response.






