
Abstract
Gastrointestinal stromal tumours (GISTs) are the most common mesenchymal tumours of the gastrointestinal tract, arising from the interstitial cells of Cajal. They are known to occur in all parts of the gastrointestinal tract from the oesophagus to the anorectum, with the stomach being the most commonly affected organ (60%). GISTs are commonly known to occur within the fifth and sixth decades of life, carry an equal predisposition between females and males, and are associated with tyrosine-protein kinase (KIT) or platelet-derived growth factor receptor alpha (PDGFRA) mutations in 85–90% of cases. Familial syndromes associated with GISTs are neurofibromatosis Type 1, Carney’s triad (gastric GIST, pulmonary chordoma, and paraganglioma), Carney–Stratakis syndrome (GIST and paraganglioma), and familial GISTs. Lesions vary in size from a few mm to >30 cm, with a median size between 5 and 8 cm. Immunohistochemical staining with KIT and DOG1 show the highest sensitivity for GISTs. While 20% of GISTs are diagnosed asymptomatically, and 10% at autopsy, 70% are symptomatic. Bleeding followed by abdominal pain and a mass growth are the most common symptoms. Forty to fifty percent of GISTs are biologically malignant. Malignant GISTs spread haematogenously to the liver and peritoneum, while lymphatic spread is rare. Risk stratification subdivides GISTs into very low, low, intermediate, and high-risk groups. Computed tomography (CT) scan is the mainstay of diagnosis, though they are often incidentally detected on endoscopy. Surgery offers the best chance of cure in resectable lesions, while tyrosine kinase inhibitors are the treatment of choice in non-resectable and metastatic GISTs. Neoadjuvant and adjuvant tyrosine kinase inhibitors increase resectability, time to recurrence, recurrence-free survival, and overall survival in GISTs.
INTRODUCTION
Gastrointestinal stromal tumours (GISTs) are the most common mesenchymal tumours of the gastrointestinal tract (GIT), arising from the interstitial cells of Cajal (ICC).1 Neuroenteric networks formed by the ICC are widely distributed within the submucosal, intramuscular (including the deep muscular plexus), and intermuscular layers of the GIT, from the oesophagus to the internal anal sphincter.2 This explains the diverse location of GISTs found within patients, with the stomach being the most common organ involved (60%).3 Other sites where GISTs can occur are the small intestine (30%), duodenum (5%), and colorectum (<5%).3 Rarer locations are the oesophagus and appendix which constitute <1% of all GISTs. Extra-GIT locations like the omentum, mesenteries, and retroperitoneum usually represent metastasis or a possible detachment of the GIST from its GIT origin, even though a small number of primary tumours are reported in these sites.4 The molecular hallmark of these neoplasms is a kinase-activating mutation in either the receptor tyrosine-protein kinase (KIT) or platelet-derived growth factor receptor alpha (PDGFRA) genes, which are present in 85–90% of reported tumours.5 Surgery offers the best chance of cure in localised disease.6 Tyrosine kinase inhibitors (TKIs), such as imatinib, offer improved survival rates in metastatic GISTs and decrease recurrence rates in patients with resectable but large tumour burden, with the stratification risk dependent on the location of the tumour and its respective mitotic number.7 The aim of this article is to provide an evidence-based review on the epidemiology, molecular genetics, pathology, clinical characteristics, investigations, and treatment of GISTs arising in the GIT.
METHODOLOGY
A comprehensive search without language restriction was undertaken using MEDLINE, Scorpus (including Embase), and Cochrane Central Register of Controlled Trials (CENTRAL), from 1st January 1985–30th June 2016. PubMed was also searched for in-process citations. MeSh terms used were ‘GIST’, ‘gastrointestinal stromal tumour’, and ‘tyrosine kinase inhibitor’. Important clinical trials were searched for on ClinicalTrials.gov and the International Clinical Trials Registry Platform Search Portal (ICTRP). Manual searches were carried out for recent articles in journals with high impact factors and reference lists in key articles. All evidence-based work was carried out in accordance with the standards published in the Centre for Reviews and Dissemination’s (CRD, University of York, York, UK, 2008) guidance for undertaking reviews in healthcare.8 Preference was given to articles of high quality and those with important observations or randomised trials.
EPIDEMIOLOGY
The annual incidence of GIST varies according to geographical location. Incidence of GIST is cited as 11–15 per million population, per year in the West.9 The incidence is slightly higher in the East, at 16–22 per million, including a relatively higher proportion of extraintestinal GISTs (10%).10,11 In the UK, the annual incidence of GIST cases is between 1.32 and 1.5 per 100,000, equivalent to approximately 800–900 new cases per year.12,13 Prevalence of GIST is estimated to be around 120 per million population.14 Risk stratification, as per the United States National Institutes of Health (NIH) criteria, estimates the prevalence at 18.5% for very low-risk, 43% for low-risk, 20% for intermediate-risk, and 18.5% for high-risk GISTs.14 GIST is most commonly known to occur in the fifth or sixth decades of life (80%), with a median age of 60 years.6,9 The disease carries an equal predisposition between males and females, though GISTs as a syndromic component of Carney’s triad (gastric GIST, pulmonary chordoma, and paraganglioma) occur more commonly in females.7
MOLECULAR GENETICS
The association of KIT mutations with GIST has been reported to be between 75% and 90% in various studies, with one showing that 88.2% of GISTs have mutations in the KIT gene with a gain of function;15 a PDGFRA mutation is present in 4.7% of GISTs.15 Both of these genes are located in the long arm of chromosome 4 and encode for homologous receptor tyrosine kinase proteins.16 Most KIT mutations involve exon 11, followed by exon 9, exon 13, and exon 17, in descending order of frequency.3 Imatinib, a TKI, acts best on exon 11 mutations, and least on exon 17 mutations where the drug is primarily resistant.3,15 PDGFRA mutations have a predilection for gastric GISTs, though duodenal GISTs are also seen with these mutations.3,17 Exons 18, 12, and 14 are mutated in PDGFRA mutations in decreasing order of frequency with the exon 18 PDGFRA D842V mutation being resistant to imatinib.3,17 Mutational analysis is crucial before starting adjuvant imatinib therapy to identify resistant genotypes that will not respond to adjuvant therapy (PDGFRA D842V mutation, neurofibromatosis Type I [NF1]-associated GISTs, and wild-type succinate dehydrogenase [SDH]-negative GISTs) and those that would need a higher dose (800 mg/day) of imatinib (exon 9 KIT mutation).18
FAMILIAL SYNDROMES
GIST is associated with familial syndromes in <5% of cases.3 NF1 is the most commonly associated syndrome, with 7% of patients developing GISTs.3,19 Both KIT and PDGFRA mutations are frequently lacking in these patients.20 There is a high association of NF1 with duodenal and small intestinal GISTs;3,21 although the majority of these tumours are benign and clinically indolent, malignant GISTs can occur.22 GISTs associated with NF1 are resistant to imatinib therapy.18
Carney’s triad typically lacks KIT and PDGFRA mutations, along with a distinctive SDH subunit B (SDHB) negativity on immunohistochemistry, with tumours exclusively occurring in the stomach.21,22 Recently, SDH subunit C (SDHC) epigenetic hypermethylation has been reported in this syndrome.23 A vast majority of these GISTs occur at a young age and carry a female preponderance (85%).3 Although the majority of these tumours are clinically benign, liver metastasis are known to occur as they can behave unpredictably, even after risk stratification.
Carney–Stratakis syndrome (GIST and paraganglioma) also typically lacks KIT and PDGFRA mutations but can carry a germline mutation in any subunit of the SDH gene.24 Tumourigenesis in this syndrome hinges upon a germline SDHB, SDHC, or SDHD mutation, coupled with a somatic inactivation of the corresponding wild-type allele in the tumour.25 SDHA mutations (30%) have also been identified on immunohistochemical analysis of a few cases.26
Familial GISTs are associated with germline mutations of either the KIT or PDGFRA genes.3,27 Transmission is autosomal-dominant, and the affected are at a high risk of developing gastric or small bowel GISTs at middle age. Other manifestations associated with familial GISTs due to germline KIT mutations include: cutaneous hyperpigmentation, mast cell disorders, diffuse hyperplasia of ICC, and dysphagia, while those due to germline PDGFRA mutations include inflammatory fibroid polyps of the stomach and small bowel, gastrointestinal lipomas, and large hands.28
PATHOLOGY
GISTs vary in size remarkably, ranging from a few mm to >30 cm in size, with the median size being between 5 cm and 8 cm.6 Micro-GISTs (<1 cm) are often found incidentally in resected specimens of gastro-oesophageal junction.29 Micro-GISTs have been found to be present in 10.0–22.5% of resected specimens and autopsy tissue, and there is evidence to suggest that these are precursor lesions to macroscopically relevant GISTs after further molecular alterations.30-32 Macroscopically relevant lesions usually show an exophytic pattern, often with compression of other intra-abdominal organs. Microscopically, GISTs fall into three basic categories: i) epitheloid; ii) spindle cell; and iii) mixed variety.14,29 Four subtypes of epitheloid and four of the spindle cell variety have been identified.3
Immunohistochemistry is often necessary to confirm the diagnosis. More than 95% of tumours stain positive for KIT (CD117) and DOG1 which are the most sensitive and specific markers for the diagnosis of GISTs.6,7 DOG1 and PKC-θ expression is especially useful to identify a subset of KIT– tumours who will respond to KIT-targeted treatment.33,34 PKC-θ has been found to have a high sensitivity but low specificity in the diagnosis of GISTs, therefore its use for routine diagnosis of these tumours is not recommended.35 Other markers included in the GIST panel are CD34 (70–80%), smooth muscle actin (30%), desmin (<5%), and S-100 (rare).34 PDGFRA staining lacks specificity, is technically challenging, and is pushed into the second panel by a few pathologists.34
CLINICAL FEATURES
On average, 70% of GISTs are symptomatic, while 20% are diagnosed asymptomatically; 10% are diagnosed only at autopsy.6 Clinical signs and symptoms depend on the site of the tumour.36 The most frequent symptoms are vague abdominal discomfort (60–70%), bleeding (30–40%), anaemia, dyspepsia, vomiting, and weight loss.6 Large tumours present with a palpable lump in the abdomen with mass effect. Bleeding may be chronic where the patient presents with chronic iron deficiency anaemia requiring blood transfusion. Acute bleeding may be intraluminal which manifests as haematemesis and melena, or extraluminal due to tumour rupture, presenting as an acute abdominal catastrophe. Site-specific symptoms include dysphagia for oesophageal GISTs, obstructive jaundice for duodenal or periampullary GISTs, and intussusception or bowel obstruction in small bowel GISTs.36 Spread occurs either haematogenously or transcoelomically to the liver, mesentery, omentum, and peritoneum. Lymphatic spread is rare, except in wild-type GISTs such as paediatric wild-type GISTs, and those occurring in the setting of Carney’s triad where nodal metastasis may occur in 20–30% of cases.37
RISK STRATIFICATION
GISTs represent a class of tumours with varied biological behaviour without a sharp distinction between benign and malignant lesions. It is difficult to predict malignancy of a GIST in the absence of metastasis due to lack of absolute histological criteria.38 It is estimated that 40–50% of GISTs are biologically malignant, and half of these show evidence of metastatic spread to the liver or peritoneum at the time of diagnosis or primary surgery.39 In a large cohort of 439 patients,40 24% of GISTs were locally advanced or metastatic, precluding curative resection. Of those resected, 45% had a recurrence at follow-up, making a total of 69% which were presumed malignant. The overall incidence of liver metastasis was 54%, and peritoneal metastasis was 62%.40 This observation suggests that a high proportion of GISTs, which are biologically malignant, do not show evidence of loco regional spread at the time of initial diagnosis. Thus, a reliable risk scoring system is thought to be needed to identify lesions that are at a high risk of recurrence and metastasis which would benefit from adjuvant TKI therapy, as well as excluding patients who would not benefit from it.
The National Institutes of Health Scoring System
The NIH Scoring System, also known as the Fletcher’s Risk Criteria, is the first risk classification system adopted for GISTs.41 This incorporates only two risk factors, namely tumour size and mitotic count per 50 high-power field. Even though GIST-specific data were not available at the time of incorporation of this system, NIH criteria are fairly accurate in identifying GISTs which are at high risk of recurrence.40 However, this system does not include tumour site or rupture as risk factors. The NIH Scoring System tends to overestimate risks in large but biologically inactive gastric GISTs, and underestimates the risks in small duodenal and rectal GISTs which can be biologically aggressive.42
The Armed Forces Institute of Pathology Scoring System
The Armed Forces Institute of Pathology (AFIP) scoring system was the first risk classification for GISTs based on evidence which included anatomic site, along with tumour size and mitotic count.39 It also seeks to establish the size of real risk expressed as a percentage.39 This system identifies the fact that gastric GISTs have an overall better prognosis than intestinal GISTs.39,43,44 By virtue of establishment of the quantum of real risk, it helps the oncologist to reliably decide the need for adjuvant TKI therapy.38 The National Comprehensive Cancer Network (NCCN) GIST Task Force adopted the AFIP scoring system within their clinical practice guidelines regarding GISTs.45
Joensuu’s Risk Criteria
Joensuu proposed a modification of the NIH criteria in 2008 (Table 1), and incorporated anatomic site (as in the AFIP scoring system) and tumour rupture as criteria for absolute high risk, irrespective of the other risk factors.46 Tumour rupture can occur either spontaneously (80%) or during surgery (20%).47 Joensuu’s criteria can be applied to all anatomical sites of tumour origin and utilises two cut-offs for mitotic count. It has been found to be particularly advantageous in predicting tumours that are at a high risk of relapse for consideration of adjuvant imatinib therapy.47
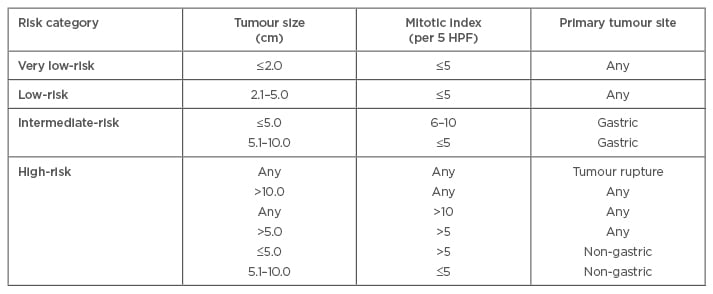
Table 1: Definition of Joensuu’s risk stratification for gastrointestinal stromal tumours.41
HPF: high-power field.
INVESTIGATIONS
Most GISTs are diagnosed either at endoscopy or by an unenhanced or contrast-enhanced computed tomography (CT) scan without a prior clinical suspicion of its presence. A quarter of these are diagnosed asymptomatically (incidental finding).7 Endoscopy may give a suspicion of GIST by detecting a submucosal lesion warranting further imaging. In a few of these cases, this turns out to be extrinsic compression by a dilated gallbladder or an enlarged spleen. The recommended imaging modalities for GISTs are contrast-enhanced CT scans, endoscopic ultrasonography (EUS), magnetic resonance imaging (MRI), and positron emission tomography (PET) scan. Our strategy is to evaluate suspected tumours following endoscopy with a contrast-enhanced CT scan of the chest, abdomen, and pelvis, as the initial imaging modality which is usually sufficient to guide further management if no metastasis is detected.
The characteristic CT appearance of a primary GIST is a large, hypervascular, and heterogeneously-enhancing mass with areas of haemorrhage, necrosis, and cystic degeneration.48 MRI is reserved as the preferred imaging modality for anorectal GISTs.49 We also use MRI to characterise suspected liver metastasis detected on CT scans. EUS is useful in detecting early primary lesions in the oesophagus, stomach, duodenum, and anorectum.50 Characteristically, they show an echo-poor pattern in the fourth layer (corresponding to the muscularis propria) or rarely in the second layer (corresponding to the muscularis mucosae) as a well-demarcated, homogenous mass. EUS-guided fine needle aspiration can be used for cytological or histological confirmation, though the characteristic sonological appearance in itself often suggests the diagnosis. EUS, unlike a CT scan, does not always offer a complete staging for metastasis even though GISTs can be metastatic at presentation in ≤50% of cases.14 PET scans are particularly useful for detecting metastatic disease and for rapid assessment of response to imatinib much before the therapeutic response to the drug can be picked up by a CT scan.51 PET scans however are not sensitive in detecting primary lesions <2 cm in size.52 A combination of PET and CT scanning (PET-CT) has shown better sensitivity, specificity, and accuracy in the staging and restaging of patients with GISTs and optimising treatment than either modality alone.53
TREATMENT
Treatment of GIST depends on resectability, site, size, and presence of metastasis and should be discussed by a multidisciplinary team which should include the relevant specialist surgeon, oncologist, radiologist, and histopathologist.
Resectable/Non-Metastatic GISTs
For resectable/non-metastatic GISTs <2 cm in size, the treatment option is tailored according to the site and risk criteria.54 All symptomatic gastric GISTs are candidates for surgical resection. Selective asymptomatic gastric GISTs without high-risk criteria on endoscopic ultrasound may be followed-up until 3 cm in size.55 For tumours in the small bowel, duodenum, or anorectum, surgical resection is advisable due to greater chances of the tumour being symptomatic and at risk of malignancy.
For tumours >2 cm in size, the treatment of choice is surgery with a 1–2 cm margin, aiming for an R0 resection.7 Organ preservation should be attempted but not at the expense of positive margins. Routine lymphadenectomy is not indicated except in those at high risk. Neoadjuvant imatinib therapy for 2–6 months has shown promise in downstaging treated tumours.56,57 Tumour shrinkage is especially useful in gastroesophageal junction tumours before gastrectomy, duodenal GISTs before pancreatoduodenectomy, and anorectal GISTs before abdominoperineal resection by facilitating achievement of R0 resection. Response to preoperative therapy is assessed by Choi criteria which has been found to be superior to the RECIST (Response Evaluation Criteria in Solid Tumors) criteria.6 Responsive tumours show a ≥10% reduction in the size of the tumour, and a 15% reduction in tumour density.6 Laparoscopic resection has been found to be safe and is advantageous in surgically amenable areas like the anterior wall of the stomach and small intestines, due to lower morbidity and shorter hospital stay.6 A combined laparoscopic and endoscopic approach (laparoscopic endoscopic-guided surgery) has been found to be an attractive option in selective gastric tumours where the tumour is transilluminated endoscopically for localisation, and resection is carried out laparoscopically.58
Adjuvant imatinib therapy has shown survival benefit in lesions of non-gastric origin, lesions >5 cm, tumour rupture, and tumours with high mitotic count.6 The usual adjuvant dose is 400 mg/day with an exception being exon 9 KIT mutations where, due to partial resistance, a dose of 800 mg/day is preferred. Imatinib therapy is not recommended in patients with resistant mutations like PDGFRA D842V, and has a doubtful role in GISTs associated with NF1 or wild-type SDH– tumours.18 Imatinib therapy is recommended for at least 3 years in patients with high-risk lesions (Table 1, Figure 1).
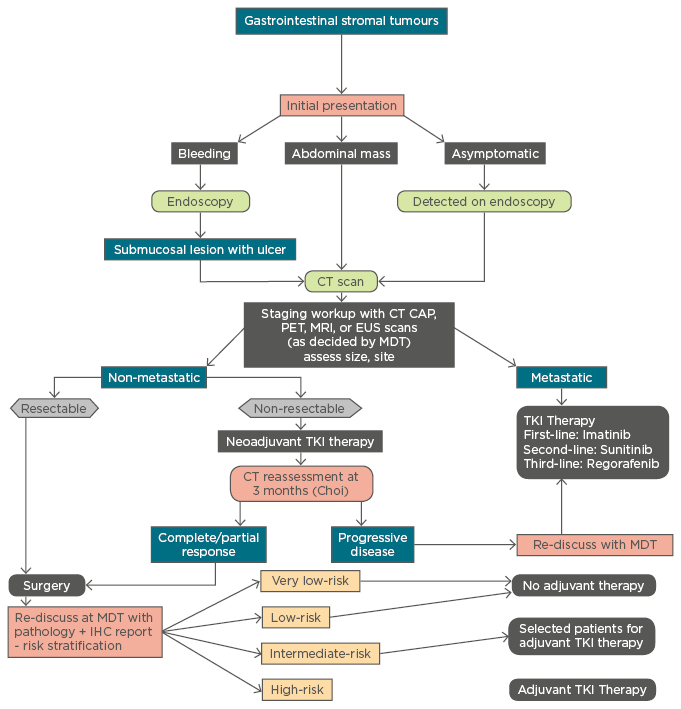
Figure 1 : Management algorithm for gastrointestinal stromal tumours.
CT: computerised tomography; CAP: chest, abdomen, pelvis; PET: positron emission tomography; MRI: magnetic resonance imaging; EUS: endoscopic ultrasound; MDT: multidisciplinary team; TKI: tyrosine kinase inhibitor; IHC: immunohistochemistry.
Non-Resectable/Metastatic GISTs
Treatment of choice for metastatic or non-resectable GISTs is TKI therapy.5,6 Imatinib is used as the first-line drug in metastatic or non-resectable GISTs at a dose of 400 mg/day, which can be increased by up to 800 mg/day in the case of non-responsiveness at lower dose or in lesions with partially resistant mutations (exon 9 KIT mutations).6,18 Sunitinib is recommended by the National Institute for Health and Care Excellence (NICE) guidelines in the UK for patients with metastatic GISTs who do not respond to imatinib.59 Regorafenib, a multikinase inhibitor, is the third drug licensed by the US Food and Drug Administration (FDA) in patients who have shown progression of the disease alongside imatinib and sunitinib therapy after successful completion of the GRID trial.60 Sorafenib, also a multikinase inhibitor, has been similarly found to be effective in imatinib and sunitinib-resistant GISTs by the Korean Gastrointestinal Stromal Tumors Group.61
The role of cytoreductive surgery in metastatic GISTs is limited and is individualised based on the tumour characteristics and performance status of the patient.6 Surgical debulking following preoperative imatinib therapy is shown to offer a survival benefit.6 The addition of adjuvant intraperitoneal chemotherapy with cisplatin and mitomycin C or doxorubicin to surgical debulking has been shown to increase the median time to recurrence from 8 months to 21 months.6 Radiofrequency ablation and liver transplantation followed by adjuvant imatinib for GIST-related liver metastasis has been attempted with promising results.62,63 A simplified algorithm for the management of GISTs is enumerated in Figure 1.
FOLLOW-UP
The follow-up strategy for GISTs varies according to the surgical culture and epidemiology of the disease in different countries. Our strategy is to follow the Association of Upper Gastrointestinal Surgeons of Great Britain and Ireland (AUGIS) Guidelines for the follow-up of GISTs.52
- Very low-risk tumours: no further imaging
- Low-risk tumours: CT at 3 months after surgery, then clinical follow-up
- Intermediate-risk tumours: CT at 3 months after surgery, then 6 monthly for 2 years, then annually up to 5 years
- High-risk tumours: CT at 3 monthly for 2 years, then 6 monthly for a further 2 years, then annual scans on an indefinite basis
- Adjuvant therapy with imatinib: CT at 3 months after surgery, then 6 monthly for 2 years, then annually up to 5 years
- Clinical suspicion of recurrence: CT
MRI can be considered as a substitute in patients who are at risk of contrast toxicity, and to reduce radiation dose from annual scans.
PROGNOSIS
Recurrence rates and survival of patients with GISTs are strongly influenced by tumour size, site, mitotic activity, tumour rupture, and extensiveness of resection.5,43,45 After resection, adjuvant imatinib therapy has been found to improve recurrence-free survival, compared to non-adjuvant therapy, from 83–98% at 1 year with no difference in terms of overall survival between the two groups monitored by the ASOCOG Z9001 trial on 713 patients with tumour size >3 cm.64 The European SSGXVIII/AIO trial which compared patients on adjuvant imatinib therapy for 3 years with those receiving the same therapy for only 1 year, found a superior recurrence-free survival (66% versus 48% at 5 years) and overall survival (92% versus 82% at 5 years) in patients who received the adjuvant therapy for 3 years.65 The recurrence-free survival and overall survival observed in the various trials on the effect of TKIs for GISTs are enumerated in Table 2.
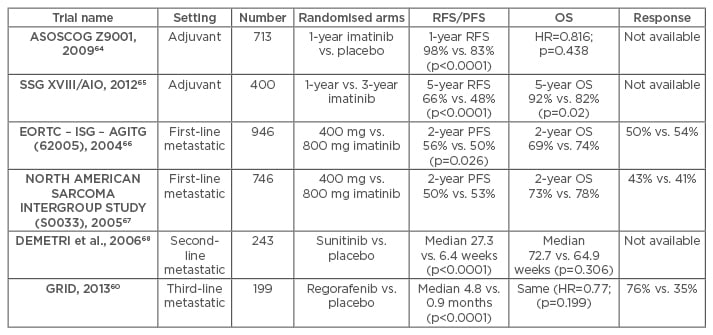
Table 2: Key trials with tyrosine kinase inhibitors in patients with gastrointestinal stromal tumours.
RFS: recurrence-free survival; PFS: progression-free survival; OS: overall survival; HR: hazard ratio.






