Abstract
Mutations in the SDHB gene cause a characteristic syndrome that includes paragangliomas (PGL) and phaeochromocytomas (PCC). Herein we present a rare case of an ependymoma in a patient with a germline SDHB mutation.
A 41-year-old man with a positive family history of PGL/PCC syndrome was found to have the familial SDHB mutation. Screening imaging for paragangliomas revealed an incidental presumed ependymoma originating from the fourth ventricle. He was followed with serial imaging to assess for progression of the lesion. Due to slow, substantial growth of the tumour, and increasing symptoms which included diplopia, unsteadiness, and wide-based gait, he underwent a resection 5 years after the lesion’s identification. Following resection of the tumour, the pathology confirmed the tumour as a posterior fossa type B (PFB) ependymoma of the fourth ventricle. Unfortunately, on Day 26 post-operatively, the patient had a pulmonary embolism and died. His family consented to an autopsy, which revealed the presence of a clinically-silent pituitary neuroendocrine tumour (PitNET).
Though ependymomas are not commonly seen in PGL/PCC syndrome, they can occur. This case represents the first molecularly-characterised ependymal tumour described in this tumour predisposition syndrome. Clinicians ought to be aware of the risk of ependymoma in patients with PGL/PCC syndrome and consider this tumour when conducting their screening and follow-up of asymptomatic SDHx mutation carriers.
Key Points
1. Individuals with a family history of paragangliomas (PGL) and phaeochromocytomas (PCC) syndromes often carry genetic mutations, such as the SDHB mutation, which can increase the risk of various tumours.2. A 41-year-old male with a known familial SDHB mutation underwent screening for PGL/PCC syndrome. During this process, an incidental ependymoma originating from the fourth ventricle was discovered. This case is the first to report a molecularly categorised ependymoma found alongside an SDHB mutation reported in literature.
3. This case suggests that ependymomas may potentially be part of the tumour spectrum caused by PGL/PCC syndrome, which could have implications for expanding screening protocols in patients with SDHB mutations.
INTRODUCTION
Succinate dehydrogenase (SDH) is a mitochondrial enzyme with a role in both the Krebs cycle and the electron transport chain. The SDH genes which encodes SDH’s four subunits have been characterised as tumour suppressor genes.1 Specifically, germline mutations in SDH genes can result in hereditary paraganglioma (PGL)/phaeochromocytoma (PCC) syndrome, where a recent study shows penetrance of 21% at 50 years old, though the rate does vary throughout the literature.2 Moreover, mutations in the SDH complex iron sulfur subunit B (SDHB) are not only associated with PGLs and PCCs, but also tumours including renal cell carcinomas, gastrointestinal stromal tumours (GIST), and pituitary neuroendocrine tumours (PitNETs).3-5
Ependymomas are relatively uncommon, constituting approximately 4% of neuroepithelial tissue tumours in those over 40 years of age. Relative to other primary malignant neuroepithelial tissue tumours, Grade 2 or 3 ependymomas have a favourable 5-year survival rate of 81.2% between the ages of 40–64 years, and 56.9% over the age of 65 years, in Canada.6
Ependymomas are primary central nervous system tumours that putatively originate from ependymal cells lining the cerebral ventricles and spinal cord.7 The clinical symptoms associated with ependymomas often include headaches, vomiting, and diplopia.8 Gold standard treatment is maximal safe surgical resection followed by radiotherapy depending on tumour location and residual and molecular features. Chemotherapy has a relatively limited role in the management of adult ependymomas.9
Ependymomas are not known to be caused by germline SDHB mutations. Herein, the authors present a case of a patient with a germline SDHB mutation who developed a posterior fossa type B (PFB) ependymoma and was also incidentally found to have a PitNET. The patient did not have PCC or PGL.
CASE PRESENTATION
A 41-year-old male presented for pre-symptomatic genetic testing. His maternal aunt was found to have an SDHB mutation and was diagnosed with tumours characteristic of PGL/PCC syndrome in her thirties. This led to the rest of the family having genetic testing. The patient was found to be a carrier of the familial mutation in the SDHB gene c.137G>A (p.Arg46Gln), which is pathogenic for hereditary PGL/PCC syndrome.10 The patient’s mother, as well as a maternal first cousin, were both also confirmed to have the SDHB mutation. Furthermore, the patient also noted that two maternal aunts had developed brain tumours; however, further details including their diagnoses or whether they were carriers of the SDHB mutation were unavailable.
The patient was started on a pre-symptomatic screening plan, which involved serial monitoring of urinary fractionated metanephrines and catecholamines, and imaging, which included MRI of the head, neck, abdomen, and pelvis; thorax CT; and oesophagogastroduodenoscopy. The initial screening imaging series showed no signs of PCCs or PGLs. However, the brain MRI, which was meant to look for a glomus jugulare, instead revealed a posterior fossa tumour measuring 27 mm × 39 mm × 47 mm, and was thought to be an ependymal tumour. The mass originated in the fourth ventricle, extending into the foramen of Magendie and foramen of Luschka bilaterally. There was mild dilatation of the cerebral aqueduct and lateral ventricle, as well as some flattening of the medulla and lower pons. Imaging also identified a small right adrenal nodule, likely a myelolipoma, and a small left adrenal presumed adenoma.
Following the MRI scan, the patient was reviewed by neurosurgery. He reported an approximate 2-year history of dizziness, predominately with neck extension and going from standing to supine quickly. However, his neurological examination revealed only subtle horizontal nystagmus on extremes of lateral gaze.
Follow-up imaging and laboratory tests were performed for 5 years. The ependymoma slowly grew and the patient’s symptoms progressed (Figure 1).
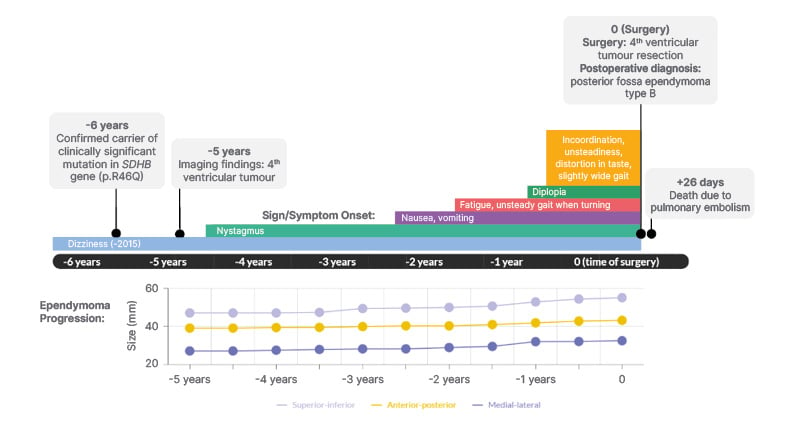
Figure 1: Timeline of significant events including symptom onset and ependymoma progression in a 41-year-old male with a germline SDHB mutation.
Ependymoma progression is reported in millimeters in the superior-inferior, anterior-posterior, and medial-lateral directions. Time 0 represents the date that the patient had surgery.
After 4 years of clinical follow-up, he developed increased dizziness with head motion, occasional nausea and vomiting in the morning, gait unsteadiness when turning, and increased fatigue. There was diplopia on left gaze and left-beating nystagmus on primary gaze and gaze-evoked nystagmus. The hydrocephalus progressed slightly and imaging revealed obvious dilation of the lateral ventricles, as well as the third ventricle.
When evaluated almost 5 years after the tumour was discovered, the patient reported hypergeusia, unsteadiness, incoordination, wide-based gait, morning nausea, and occasional vomiting. On examination, the patient was unable to perform the tandem gait test and demonstrated dysmetria on the left side. Imaging revealed that the mass was filling the fourth ventricle, extending caudally through the foramen of Magendie (Figure 2) and measured 32 mm x 43 mm x 55 mm. Given the increase in symptoms, at this point, the patient was referred to surgery with the goal for treatment to decrease symptoms and prevent progression.
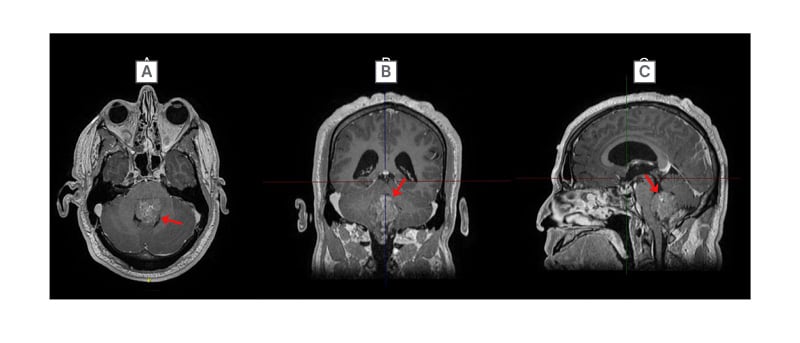
Figure 2: Final preoperative post-gadolinium T1 MRI imaging of the head in A) axial; B) coronal; and C) sagittal orientations.
Ependymoma (red arrow) filling the fourth ventricle.
He underwent a suboccipital craniotomy and C1 laminectomy for tumour resection approximately 5 years after the tumour was first discovered on MRI imaging. The pathology revealed a PFB ependymoma of the fourth ventricle and was classified as World Health Organization (WHO) Grade 2. Immunohistochemistry was used to characterise the tumour as a PFB ependymoma and showed that H3K27me3 nuclear expression was retained (Figure 3G), H3K7M mutation was immunonegative, and the Ki-67 index was 1–3%. Other immunohistochemical markers are seen in Figure 3. The postoperative MRI showed an apparent gross total resection, as well as bilateral inferomedial cerebellar infarcts. Fourteen days postoperatively, the patient had ongoing diplopia. There were bilateral abducens nerve palsies (worse on the left) and horizontal nystagmus, which likely stemmed from a perioperative cerebellar ischemic stroke. Otherwise, the patient moved his extremities well and was considered stable from a neurological perspective. There were plans for a staging lumbar puncture and an MRI complete spine.
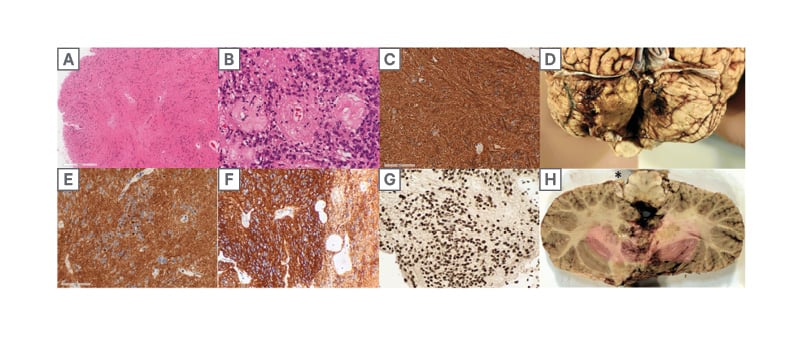
Figure 3:
A, B) The tumour consists of uniform round-to-ovoid cells in a fibrillary matrix with perivascular pseudorosettes (H, E).
C) immunohistochemistry for glial fibrillary acidic protein demonstrates diffuse strong cytoplasmic expression in tumour cells.
D) posterior view of infratentorial area with bilateral cerebellar infarcts.
E, F) diffuse cytoplasmic immunopositivity for D2-40 (D) and CD99 (F), with occasional dot-like perinuclear expression.
G) H3 K27me3 expression is entirely retained in tumour cell nuclei. H) axial slice of the rostral medulla and cerebellum with residual tumour (*) at the left foramen of Luschka.
While rehabilitating in hospital, 26 days postoperatively, he collapsed suddenly and could not be resuscitated. The patient had been receiving pharmacological deep vein thrombosis prophylaxis. An autopsy was performed, which confirmed a saddle pulmonary embolism as the cause of death.
Bilateral cerebellar infarcts were also visualised during the autopsy (Figure 3D) and the patient was also found to have residual ependymoma in the foramina of Luschka (Figure 3H) and subarachnoid space. There was also a pituitary neuroendocrine tumour, which was 5 mm in size. This tumour was of somatotroph subtype, Pit-1 positive, growth hormone expressing, and densely granulated (immunohistochemical markers seen in Figure 4). No phaeochromocytomas or paragangliomas were identified at autopsy.
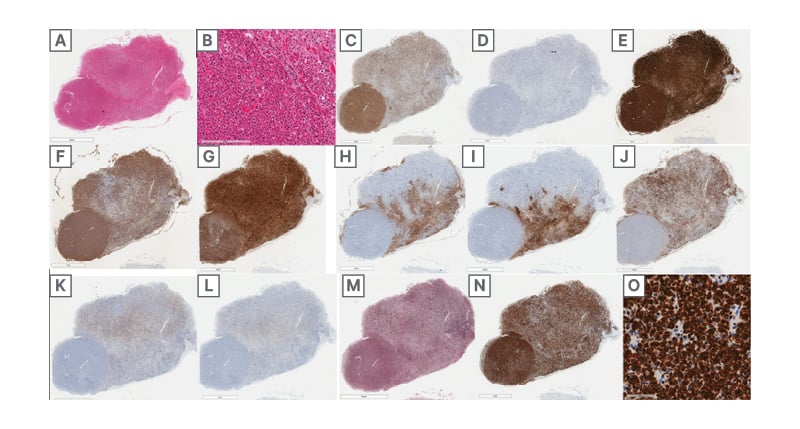
Figure 4:
A,B) H&E section of pituitary gland with a round well-circumscribed hypercellular lesion (A) featuring tumour cells (B) with predominantly eosinophilic cytoplasm, densely-arranged in sheets (B, left) relative to the adjacent uninvolved pituitary (B, top-right).
C,D) tumour cells are immunopositive for the pituitary lineage factor Pit1 (C), while being immunonegative for SF1 (D).
E) strong synaptophysin expression is supportive of a pituitary adenoma/pituitary neuroendocrine tumour.
F,G) tumour cells diffusely express growth hormone (F) and α-hCG (G).
H,I,J,K,L) expression of adrenocorticotropic hormone (H), thyroid-stimulating hormone (I), prolactin (J), luteinizing hormone (K), and follicle-stimulating hormone (L) is restricted to the uninvolved pituitary gland.
M) reticulin silver stain demonstrates a loss of normal acinar architecture in the tumour.
N,O) tumour cells diffusely express cytokeratin 8/18 stronger than background pituitary (N), with rare fibrous
bodies (O).
DISCUSSION
Mutations in the SDH genes are associated with specific PGL syndromes. These syndromes are inherited in an autosomal dominant fashion and have characteristic clinical features depending on the SDH mutation present.3 Notably, ependymomas are not commonly seen with any of the SDHx mutations, and from the authors’ literature search, up to June 2024, there has only been one report of an SDHD mutation alongside an ependymoma.11 The study examined the impact of higher altitudes on the development of more severe PGL syndrome 1 (PGL1), and unfortunately, no details were provided about the patient that developed the ependymoma. While the occurrence of concurrent PGLs/PCCs and ependymomas is relatively uncommon, it has been reported previously; however, these did not occur alongside SDHx mutations. One occurrence had an unknown genetic status, and the other occurred alongside heritable multiple endocrine neoplasia type 2A caused by a D631YRET mutation.12,13 The authors have presented a patient with an ependymoma and SDHB mutation, and to their knowledge, following a literature conducted in June 2024, this combination has not otherwise been reported in the literature.
Specifically, mutations in the SDHB gene have been associated with PGL syndrome 4 (PGL4). Compared to other syndromes, PGL4 generally presents with higher rates of PCCs, thoracoabdominal PGLs, renal cell carcinomas, and distant metastases. Furthermore, like the syndromes occurring with SDHD mutations (i.e., PGL1) and SDHA mutations (i.e., PGL5), PGL4 is also associated with GISTs and pituitary neuroendocrine tumours. In contrast, relative to many other PGL syndromes, PGL4 is associated with lower rates of head and neck PGLs, and lower rates of multifocal disease.3 The patient in the authors’ article did not present with many of the characteristic neoplasms of PGL4. Furthermore, the specific growth hormone-secreting pituitary neuroendocrine tumour that the patient presented with can cause acromegaly, which has been associated with a hypercoagulable state, and as a result, massive pulmonary embolisms.14 Interestingly, whilst the patient had a positive family history of PCCs/PGLs, he also had a positive family history for other brain tumours. Although ependymoma has only been reported with a SDHx mutation once, the relationship between SDHx mutations and other tumours, including ependymomas, has been explored, as SDH genes have proposed roles in tumourigenesis in general, and there are occasionally reports of various non PGL syndrome–like tumours occurring in those with SDHx mutations.15
The SDH gene has been suggested to have a possible genotype–phenotype–environment relationship on PGL syndromes. This has been explored by Astrom et al.,11 who have reported the only other occurrence of an ependymoma alongside an SDH mutation. The influence of higher altitude on PGL1 disease (i.e., those with SDHD mutations) was explored given the increased incidence of PGLs found in those living at high altitudes and the hypothesis that mutations in SDHD disrupt oxygen sensing in the carotid bodies, which may result in tumour formation. They found that, indeed, high altitude plays a negative role in the PGL1 phenotype, wherein those at higher altitudes had, for example, an increased number of tumours at first diagnosis and had symptoms at earlier ages. A number of the participants in this study were also reported to have developed non-paraganglionic tumours, including an ependymoma; however, these non-paraganglionic tumours did not have an association with altitude.11
Because SDH genes have role in DNA hypermethylation and hypoxia-inducible factor activation modification, their role in tumourigenesis has been explored.16,17 It has been demonstrated that tumours associated with SDHB mutations, such as GISTs, stain negative for SDHB immunohistochemistry; however, if the GIST was sporadic, in most cases there was positive SDHB immunohistochemistry.18 In studies that have assessed SDHB immunohistochemistry in ependymomas, SDHB expression was preserved, suggesting SDHB did not play a role in the tumour’s genesis.19 These findings were further corroborated by another study which found that ependymomas had preserved SDHB expression, though other central nervous system tumours, such as hemangioblastomas, did show loss of SDHB expression.20 Taken together, while SDHB expression may be altered in some tumours, expression is normal in most ependymomas, suggesting that SDHB may not play a large role in formation of ependymomas.
In 2021, a consensus statement was published, outlining recommendations for screening and follow-up of asymptomatic carriers of SDHx mutations. Recommendations for screening include blood pressure monitoring; an assessment of possible symptoms and signs of a jugular foramen paraganglioma (e.g., dysphagia, palpitations, and neck mass); measure of urine or plasma levels of metanephrines and normetanephrines; an MRI of the head, neck, abdomen, and pelvis; and a PET-CT. Given the continued risk of developing PCCs/PGLs, it has been suggested that individuals should have a regular follow-up following a negative workup, with an annual blood pressure measurement, biennial plasma or urine metanephrines, and an imaging series every 3 years.21
CONCLUSION
The authors present a unique case of a patient with an incidentally discovered Grade 2 PFB ependymoma and a small PitNET in the context of a known germline, pathogenic SDHB mutation. The patient did not have PCCs or PGLs, which are often seen in the syndrome. Although there is a chance the ependymoma was unrelated to the SDHB mutation, based on this report and one other report in the literature, the authors suspect that the spectrum of tumours caused by PCC/PGL syndrome includes ependymomas too.






