Abstract
Immunotherapy using chimeric antigen receptor (CAR)-engineered T cells has encountered important limitations in the transition of their use from liquid to solid tumours. Success is dependent upon T-cell trafficking to, and functional persistence within, tumours that often present a metabolically and immunologically hostile microenvironment. Moreover, CAR targets that are tumour specific are extremely scarce. To address these issues, several strategies have been proposed to improve both tumour selectivity and safety. One approach involves the engineering of CAR-T cells that only deploy their effector function at tumour sites. Conceptually, a solution for this exploits the oxygen-limited nature of advanced tumour deposits through the engineering of CAR that are exclusively expressed or activated under conditions of profound hypoxia. T cells have a complex inter-relationship with oxygen, which also needs to be factored into the refinement of these technologies. Ideally, oxygen-sensing CAR should only function when oxygen tension is below 2%, as is commonly the case in solid tumours but rare in healthy tissue. Successful advancement of such technologies presents opportunities for solid tumour immunotherapy because it should broaden the target repertoire that may safely be exploited in this context.
INTRODUCTION: THE CHALLENGE OF SOLID TUMOURS
Chimeric antigen receptors (CAR) are fusion molecules that couple the binding of a native tumour-associated target to the delivery of a bespoke T-cell activating signal. To extend the success of CAR-T cells from the haematological to the solid tumour setting, a number of key challenges have hindered progress.1Firstly, there are numerous physical constraints that hinder CAR-T cell recruitment, activation, proliferation, and persistence within solid tumour deposits. Secondly, CAR-T cells that do manage to navigate into the solid tumour microenvironment must then operate in the face of the adverse metabolic conditions at the site, notably hypoxia, low pH, elevated lactate concentration, and deprivation of nutrients such as glucose and amino acids.2 Even in the setting of more amenable haematological malignancies such as B-cell leukaemias and lymphomas, important safety concerns include potentially lethal cytokine release syndrome and neurotoxicity. In solid tumours, the relative absence of truly tumour-selective targets means that on-target, off-tumour toxicity is a further major concern that has caused morbidity and death of some patients.3
Numerous strategies have been developed to improve the therapeutic index of CAR-T cell immunotherapy.4,5These include the intrinsic modification of CAR architecture, engineering of systems to control CAR-T cell persistence, and use of loco-regional delivery systems to minimise systemic exposure to these cells.6,7Exemplifying this, a recent clinical trial has demonstrated the safety and efficacy of mRNA-transfected c-Met-CAR-T cells following their intratumoural administration, thereby achieving transient expression of a potentially toxic CAR primarily at the site of disease.8 Similarly, intratumoural delivery of panErbB CAR-T cells has been safely employed in the treatment of patients with relapsed or refractory and locally advanced head and neck cancers.9 More complex strategies that set out to ensure safety include the use of suicide genes,10 inhibitory CAR,11 dual antigen-targeted systems,12 combinatorial SynNotch CAR,13 and switchable CAR.14-17 To maximise success and safety requires a system that tightly restricts the activation of CAR-T cells to the solid tumour microenvironment.
In 2019, the Nobel Prize in Physiology or Medicine was awarded jointly to William Kaelin, Peter Ratcliffe, and Gregg Semenza for their work which characterised how cells sense and adapt to ambient oxygen levels. Given the profoundly hypoxic nature of many solid tumours, this paper considers the potential effects of hypoxia on CAR-T cells and how these may be harnessed to enhance the tumour specificity of these cells.
HYPOXIA OF THE TUMOUR MICROENVIRONMENT
Hypoxia refers to a low concentration of oxygen and is a frequent attribute of solid tumours.18 It commonly results from rapid tumour growth that outstrips an inadequate and often abnormal blood supply. Hypoxia has been associated with tumour cell invasiveness, metastasis, epithelial to mesenchymal transition, maintenance of cancer stem cells, and resistance to both radiation and cytotoxic chemotherapy.19-21 While oxygen tension in normal tissues is generally ≥5% and reaches approximately 14% in the lung, levels fall to below 2% in solid tumours and in chronically inflamed tissues.22 Oxygen plays a fundamental role in many metabolic processes and cells must adapt nimbly to fluctuating levels of this nutrient in order to remain viable.23-25 The key regulators of oxygen homeostasis are hypoxia-inducible factors (HIF): HIF-1, HIF-2, and HIF-3.26 While the importance of HIF-3 is less apparent, HIF-1 and HIF-2 operate as transcriptional regulators of an overlapping set of genes that mediate the hypoxic adaptive response.27
HIF are heterodimers comprising a stably expressed β subunit and a hypoxia-dependent α subunit. At normal oxygen levels, HIF-1α is rapidly degraded. This process is dependent upon the hydroxylation of proline residues found in the oxygen-dependent degradation domain (ODD) of HIF-1α by the enzyme, prolyl hydroxylase. Upon hydroxylation, the von Hippel–Lindau protein (VHL) binds and orchestrates HIF-1αubiquitination followed by proteasomal degradation.26 Conversely, under hypoxic conditions, the HIF-1αprotein accumulates, translocates to the nucleus, and dimerises with the HIF-1β subunit, forming the HIF-1 complex. The latter can then bind to hypoxia-responsive elements in the promoter region and increase transcription of specific hypoxia-induced genes.28
It has been argued that hypoxia represents the best validated cancer-associated feature that has yet to be exploited clinically.29 A wide number of experimental treatments are focussed on harnessing tumour-associated hypoxia, including prodrugs30 and HIF inhibitors.31 CAR-T cell immunotherapy strategists also need to consider the challenge and potential opportunity presented by the profoundly hypoxic nature of many solid tumours. Recent studies have investigated the effects of oxygen deprivation on CAR-T cells and some research groups have gone further to exploit hypoxia by developing oxygen-sensing CAR-T cells.
CAR-T CELLS AND HYPOXIA
Little is known about how CAR-T cells behave in an hypoxic environment. However, hypoxia is known to exert several effects on unmodified T cells, including the induction of apoptosis, compromised activation, and upregulated expression of inhibitory receptors.18,32 Nonetheless, there is evidence that these inhibitory effects of hypoxia are not generalisable for all T-cell subsets. It has been argued that hypoxia favours the differentiation of Th9, Th17, and Th22 cells while hindering the differentiation of Th1 and cluster of differentiation-8 (CD8) effector T cells, while data on the effect on regulatory T cells are conflicting.18,33Moreover, Xu et al.34 showed that hypoxia enhanced the proliferation, survival, and cytolytic activity of effector memory T cells, in contrast to its inhibitory effects on naïve and central memory T cells. This effect was attributed to elevated glycolytic activity, accompanied by enhanced HIF-1α expression. Hypoxia enhanced the cytotoxic activity of a CAR directed against the GD2 ganglioside, associated with increased degranulation of the CAR-T cells upon target recognition.34 Furthermore, antigen-specific CAR-T cells that were mobilised into hypoxic tumour areas were more proliferative than CAR-T cells in normoxic areas, when evaluated in two in vivo models.34,35
Berahovich et al.36 also explored the effects of hypoxia on CAR-T cells targeted against B-cell maturation antigen (BCMA) and CD19. They observed that hypoxia diminished the expansion of both CAR- and non-transduced T cells.36 Differentiation of both BCMA and CD19 CAR-T cells was also affected, switching from central memory towards an effector memory phenotype. They also observed an increase in the CD4:CD8 CAR-T cell ratio, suggesting that CD8+ CAR-T cell expansion is more dependent on oxygen.36 While cytolytic activity of CAR-T cells that were expanded in 5% oxygen was increased, these cells manifested a significant reduction in the production of interferon-γ, IL-2, IL-6, and granzyme B, with programmed cell death protein 1 levels unaffected compared to CAR-T cells expanded in atmospheric oxygen levels.36 Consistency of findings between CD19 and BCMA-targeted receptors suggests that the effects of hypoxia were CAR-independent.
These in vitro data provide an insight into the way CAR-T cells function in a microenvironment restricted of oxygen, simulating that found in the tumour microenvironment. These findings also provide the basis for design of advanced CAR systems that actively exploit tumour-associated hypoxia for therapeutic gain.
RESTRICTING CAR-T CELL FUNCTION TO HYPOXIC ENVIRONMENTS
The first attempt to exploit the hypoxic tumour microenvironment to regulate CAR expression was reported by Ang et al.37 In an abstract publication, they described the use of the ‘Sleeping Beauty’ transposon system to express a second generation (CD28+CD3ζ) anti-CD19 CAR fused to an ODD to restrict CAR expression to conditions of hypoxia. No expression of the ODD-containing CAR construct was seen at atmospheric oxygen (referred to as normoxia hereafter), while cell-surface CAR expression was detectable following the transfer of cells to 1% oxygen.37
The following year, this group described an oxygen-sensitive c-Met-specific CAR that utilised a fused ODD domain derived from HIF-1α (Figure 1A–B).38 Like its predecessor, this CAR was expressed using the Sleeping Beauty transposon system. Cell-surface expression of the CAR and cytolytic activity against c-Met-engineered target cells were both strictly dependent upon hypoxic conditions.39 In a related approach, the clinical-stage biopharmaceutical company, Cellectis, Paris, France, described a method to create a multi-input signal-sensitive T cell. The method involved the application of a logic AND gate whereby two independent stimuli as input signals are required for CAR activation to occur. To exploit hypoxia as one such input signal, the CAR was engineered to contain an ODD. By this means, cognate ligand engagement was restricted to conditions of hypoxia.40
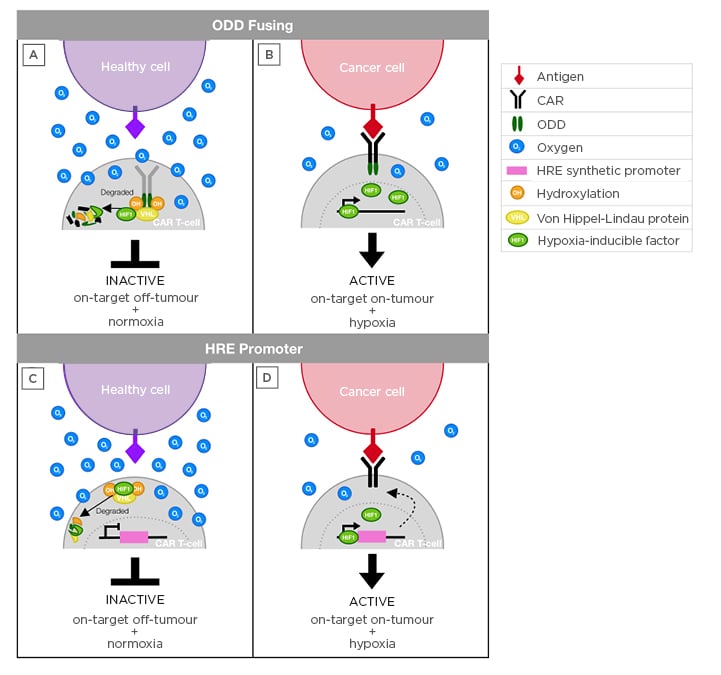
Figure 1: Schematic to illustrate hypoxia-sensing CAR-T cell approaches.
A,B) ODD fusion approach. Upon infusion, CAR-T cells may engage healthy (A) or cancer (B) cells that express cognate target. When the CAR is exposed to normoxia, the ODD fused to the CAR become hydroxylated and recognised by the VHL, which ubiquitinates the CAR for proteasomal degradation. In hypoxia (B), prolyl hydroxylase does not hydroxylate the CAR, which prevents degradation and allows target-dependent CAR activation.
C,D) HRE-promoter approach. Upon infusion, CAR-T cells situated in healthy tissue (C) are exposed to normoxia and thus CAR transcription is not triggered, protecting healthy cells that express the antigen and avoiding on-target, off-tumour toxicity. By contrast, HIF-1 complex accumulation occurs selectively in conditions of hypoxia (D), allowing the binding of this complex to HRE in the synthetic vector promoter region. As a result, CAR expression occurs, which enables the activation of these T cells upon antigen binding.
CAR: chimeric antigen receptor; HIF-1: hypoxia-inducible factor 1; HRE: hypoxia-responsive elements; ODD: oxygen-dependent degradation domain; VHL: von Hippel–Lindau protein.
The hypoxia-exploiting CAR described in this paper had a multichain (mc)CAR architecture (Figure 2). The latter was based on the high-affinity IgE receptor (FcεRI),41 a configuration that had earlier been designed to regulate CAR activity in a small molecule-dependent manner.14 The mcCAR was engineered by replacing the extracellular domain of the α chain for a single-chain variable fragment fused to a hinge domain from the CD8α chain. The native domain of the β chain was substituted with a 4-1BB domain while the CD3ζchain was appended to the γ chain endodomain. Three CAR were designed as follows. HIF-CAR1 was engineered by fusing the ODD sequence (amino acids 380–603) of HIF-1α to the α chain of the mcCAR, thereby conferring oxygen-dependent stability on the fusion receptor. In HIF-CAR2, the N-terminal domain containing the key proline residue P402 was added (amino acids 344–417), while in HIF-CAR3, the C-terminal domain containing the key proline residue P564 (amino acids 530–652) was added. As indicated above, these residues have previously been shown to interact with the VHL tumour-suppressor protein and to be hydroxylated in conditions of physiological normoxia, ultimately leading to protein degradation.42-44
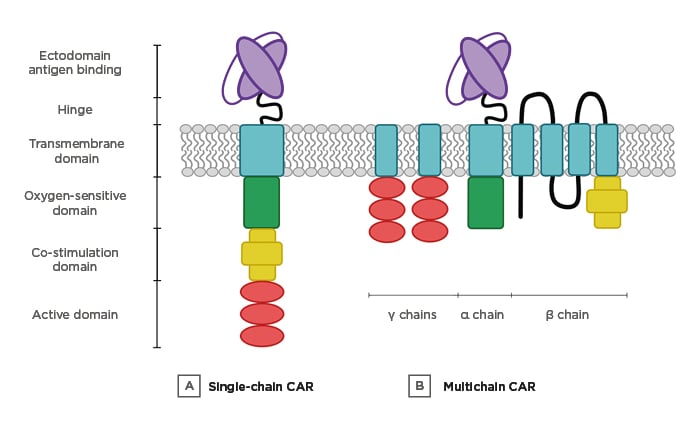
Figure 2: Multi-input CAR designs.
A) Single-chain CAR. The targeting, activation, co-stimulation, and ODD domains are fused in a unique chain. B) Multichain CAR. The different domains are distributed in three different chains (α, β, and γ).
CAR: chimeric antigen receptor; ODD: oxygen-dependent degradation domain.
To demonstrate that oxygen could be used to turn on the switch, T cells were electroporated with mRNA encoding for the CAR-ODD for comparison with a control CAR that lacked an ODD. After incubation for 20 hours under hypoxic conditions, cell-surface expression of HIF-CAR1 and HIF-CAR2 were enhanced, while the HIF-CAR3 (for unclear reasons) and the control CAR responded minimally. To demonstrate that the switch was reversible, T cells were transferred from hypoxia to normoxia for 6 hours. Cell-surface expression of HIF-CAR1 and HIF-CAR2 were both successfully negated, whereas no change in expression was observed for the control CAR.
To further evaluate the dynamics of the system and the rate of CAR decay upon removal from hypoxia, T cells were electroporated and placed in normoxia for 6 hours to recover. Next, hypoxia was applied for 16 hours and cell-surface CAR expression was monitored over 6 hours of reoxygenation thereafter. Cell-surface CAR expression decreased by 80% with a lapse of 2 hours for both HIF-CAR1 and HIF-CAR2 and this remained stable for the next 4 hours. In concordance with previous experiments, control CAR and HIF-CAR3 did not respond upon reoxygenation.
To assess cytolytic activity, control or HIF-CAR1 T cells were co-cultured at different effector-to-target ratios for 16 hours with Daudi cells (a human Burkitt lymphoma B-cell line), either in normoxic or hypoxic conditions. Tumour cell killing was observed in both hypoxic and normoxic conditions at effector-to-target ratios of 5:1 and 10:1, respectively. While enhanced killing was seen in hypoxic conditions, leakiness of the system was suggested by the detection of cytotoxic activity under conditions of normoxia.40
The strategies described thus far have taken advantage of the HIF master orchestrators of oxygen responsiveness, through the fusion of an ODD to the CAR endodomain (Figure 1A-B). An alternative oxygen-sensing CAR system was recently presented by Sarén et al.45 in an abstract. They fused a cassette of hypoxia-responsive elements to a minimal cytomegalovirus promoter and evaluated the ability of this putative hypoxia-responsive promoter to drive expression of a CD19-specific CAR (Figure 1C-D). Following chemical induction of hypoxia, they showed that expression of the CAR was enhanced, whereas the expression of a control CAR under the transcriptional control of a constitutive EF1α promoter was unaltered. Additionally, the group reported that the oxygen-sensing CAR displayed enhanced activation and cytotoxic activity upon antigen recognition in conditions of hypoxia, when compared to normoxia.45
CONCLUSIONS AND PERSPECTIVE
The aforementioned studies provide proof of concept as to how hypoxia may be exploited to enhance precision targeting of CAR-T cell immunotherapy.
A further theoretical advantage relates to the fact that restricted CAR expression at sites of hypoxia might be expected to mitigate against the deleterious effects of tonic signalling, which is a particular problem with some single-chain variable fragment-targeted CAR.46 Moreover, T cells that escape from the tumour microenvironment should be rapidly disarmed owing to CAR ubiquitination under oxygen-replete conditions. By this means, off-tumour on-target toxicity should be minimised. It should be acknowledged, however, that ODD-dependent CAR degradation can take between minutes and hours47 meaning that some such toxicity might nonetheless occur.
In the further advancement of these technologies, a number of points warrant consideration. While physiological oxygen levels vary across many normal tissues, they are lower than atmospheric oxygen levels. This highlights the need for careful testing of CAR expression and function across a range of oxygen tensions. Another potential concern is that the activity of hypoxia-sensing CAR could be suboptimal in tumours with heterogeneous oxygen levels, owing to excessive CAR degradation in physiological normoxic areas.47 Finally, leakiness of some systems could prove problematic in the clinical application of this technology. While it has been argued that low-level CAR expression under oxygen-replete conditions could help initiate CAR-T cell activation,40 concerns remain that on-target, off-tumour toxicity could occur as a result. Additionally, inflammation triggered by low-level CAR activation might be expected to cause further local hypoxia, creating a vicious circle in which further CAR upregulation, tissue damage, and inflammation could ensue. By contrast, systems that rely exclusively on HIF-triggered CAR transcription may be undermined in part by the fact that TCR activation can also trigger HIF stabilisation via mTOR signalling.48
In summary, highly stringent restriction of CAR expression to the tumour microenvironment constitutes a desirable AND gate by which to restrict CAR function at the site of disease (e.g., where target and hypoxia colocalise). However, with currently available systems, this remains an unmet goal that requires further technological refinement. The successful development of such a technology would broaden extensively the range of targets and cell dose levels that could be safely employed in efforts to develop effective CAR-T cell immunotherapies for solid tumours.






