
Abstract
Multiple myeloma (MM) is a plasma cell disorder that represents almost 10% of haematologic malignancies. Renal impairment, one of the most common complications of MM that occurs in 20–50% of patients, can present in a variety of forms and is associated with increased mortality. Myeloma cast nephropathy is the most common cause of kidney disease in MM patients, presenting as acute kidney injury in the majority of patients. The recent introduction of new chemotherapy agents, autologous stem cell transplantation, and the development of novel techniques of light chain removal have been associated with improved renal and patient outcomes in MM patients. Nevertheless, dialysis-dependent patients with MM have higher mortality than other dialysis patients and may be considered for kidney transplantation only if sustained remission has been achieved and sustained for at least 3 years, bearing in mind the risk of disease recurrence.
The authors review the most frequent renal manifestations associated with MM, namely myeloma cast nephropathy, light-chain amyloidosis, and monoclonal immunoglobulin deposition disease, focussing on the therapeutic options for acute and chronic kidney disease.
INTRODUCTION
Multiple myeloma (MM) is a plasma cell disorder characterised by the presence of >10% of clonal plasma cells in the bone marrow or biopsy-proven plasmocytoma and at least one of the myeloma-defining events (Table 1).1 MM represents 1% of all malignancies and almost 10% of haematologic malignancies.2 It is more common in elderly patients with a median age of diagnosis of 70 years, males, and African-Americans.1,3 Advances in therapy in the last decade have changed the management of this disease and prolonged patient survival.4,5
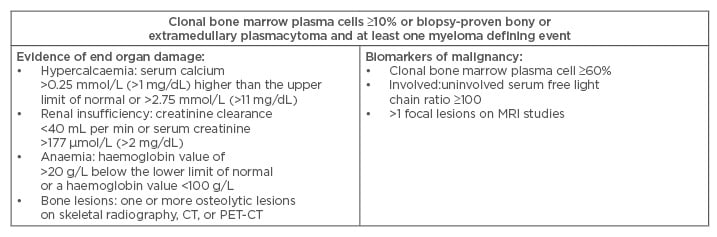
Table 1: Diagnostic criteria for multiple myeloma according to the International Myeloma Working Group.
CT: computed tomography; PET-CT: positron emission tomography–computed tomography; MRI: magnetic
resonance imaging.
Renal impairment (RI) is one of the most common complications of MM; it occurs in 20–50% of patients and is associated with increased mortality.6 It is defined as an elevated serum creatinine (>2 mg/dL) or reduced creatinine clearance (<40 mL/min), as a result of MM.6 It is important to note the increasing number of cases of RI caused by a monoclonal immunoglobulin (Ig) secreted by a clonal plasma cell disorder that does not meet the criteria for MM, but is consistent with monoclonal gammopathy of undetermined significance. These cases were thus defined as monoclonal gammopathy of renal significance.7
Renal disease in MM patients can present in a variety of forms that can be grouped as Ig-dependent, as in the causes that result from the toxic effects of monoclonal light chains, and Ig-independent (Table 2).8 The toxic effects of monoclonal light chains can affect different segments of the kidney by different mechanisms, which results in a variety of clinicopathologic syndromes, the most common being myeloma cast nephropathy (MCN).9 Renal biopsy has an important role in the evaluation of MM patients, as different renal lesions have different therapeutic and prognostic implications.9
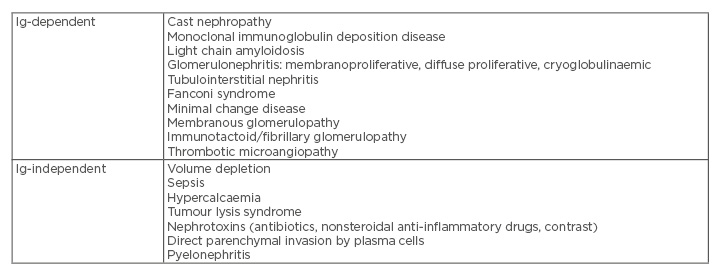
Table 2: Causes of renal disease in multiple myeloma.
Ig: immunoglobulin.
PATHOGENESIS
Monoclonal light chains are freely filtered at the glomerulus and endocytosed and catabolised by the proximal renal tubule cells.9,10 In MM, the increase in the levels of monoclonal light chains may exceed the absorptive and catabolic capacity of the proximal renal tubule cells.9,10 In the distal nephron, the light chains interact with Tamm–Horsfall protein, also named uromodulin, produced by the thick ascending limb of the loop of Henle cells, and form tubular casts, leading to tubular obstruction and inflammation. Cast formation is promoted by hypercalcaemia, dehydration, and nephrotoxins.9,11
In the proximal tubule, light chains induce direct toxic effects on proximal tubule cells inhibiting substrate transport, leading to proximal tubulopathy, also named Fanconi syndrome, and promote tubular cell apoptosis and epithelial–mesenchymal transition, leading to tubulointerstitial fibrosis.11-13
Glomerulopathy is caused by the deposition of Igs in the amyloid or non-amyloid forms.9,14 When mesangial cells and macrophages endocytose amyloidogenic light chains and other precursor proteins into lysosomes, amyloid fibril formation takes place.14 The interaction of fibrogenic monoclonal light chains with mesangial cells induces a fibroblastic phenotype and formation of extracellular matrix.14,15
As previously stated, the pathogenesis of MM-associated renal disease also includes Ig-independent mechanisms, which include hypercalcaemia, hyperuricaemia, hypovolaemia, sepsis, rhabdomyolysis, non-steroidal anti-inflammatory drugs, renin–angiotensin system inhibitors, bisphosphonates, and, rarely, pyelonephritis or direct renal parenchymal invasion by plasma cells.8
Hypercalcaemia is the second most common cause of RI in MM, impairing renal concentrating ability, causing vasoconstriction of renal vasculature, and enhancing diuresis, resulting in hypovolaemia and acute tubular necrosis.8 Hypovolaemia, sepsis, and rhabdomyolysis are also frequent causes of acute tubular necrosis in MM patients.8
Myeloma Cast Nephropathy and Acute Kidney Injury
MCN is the most common cause of kidney disease in MM patients.16 Acute kidney injury (AKI) is the most common presentation for MCN, is often associated with dehydration, infection, hypercalcaemia, hyperuricaemia, or nephrotoxins,17 and in most cases occurs in patients with serum light chains level >100 mg/dL.16 Proteinuria, primarily light chain proteinuria, is a presenting feature in all MCN patients; however, only 10% have nephrotic syndrome.17
Histologically, MCN is characterised by the presence of light chain casts in the distal tubules and collecting ducts, with immunofluorescence staining for a single light chain, and crystalline structure on electron microscopy.16,17 Also common is a giant cell reaction around the casts, tubular injury, and interstitial inflammation, which may vary from minimal to severe and are dependent on the severity and duration of obstruction.17 MCN can also coexist with other kidney lesions, such as monoclonal immunoglobulin deposition disease (MIDD) and light chain amyloidosis (AL amyloidosis).17
AKI requiring dialysis is seen in ≤10% MM patients, of which 90% have MCN.18 Hypercalcaemia is the second most frequent cause of AKI in MM patients, and can also predispose to cast formation.19 Renal insufficiency is associated with higher morbidity and mortality, and is the second most common cause of death in MM patients, after infection, thus emphasising the importance of an early and aggressive treatment,17,18 because recovery of renal function is associated with increased survival.20
Treatment requires elimination of the precipitating factors that favour cast formation, such as correction of hypercalcaemia and dehydration and increasing urine flow, and rapid reduction of serum light chain levels by chemotherapy and extracorporeal removal.17,18 The benefit of urine alkalinisation is uncertain and might increase the risk of calcification, particularly if hypercalcaemia is present.16
The management of myeloma has evolved in the last decade. The current first line of therapy is dexamethasone in combination with bortezomib or with thalidomide, cyclophosphamide, and doxorubicin, followed by autologous stem cell transplantation (ASCT).3 ASCT has higher response rates and better survival than standard chemotherapy.17,21 However, the effects of ASCT on renal response and independence of dialysis are variable.3
There is only benefit in the extracorporeal removal of light chains when it is associated with effective chemotherapy.18,22 The largest randomised study in patients with MM and AKI failed to demonstrate benefits of plasma exchange on patient and renal outcomes.22,23 The current focus is on high cut-off haemodialysis (HCO-HD), which has greater efficacy for light chain removal than plasma exchange.24 In fact, the combination of HCO-HD and chemotherapy has been associated with sustained decrease of serum light chains and high incidence of dialysis independence.25-27 Furthermore, the MYRE trial demonstrated a higher renal recovery rate with intensive HCO-HD when compared with conventional high-flux dialysers, in MM patients with MCN and severe AKI treated with a bortezomib-based regimen.28 The results of the EuLITE trial will provide further insight on the effects of HCO-HD on patient and renal outcomes.29
Chronic Kidney Disease
Chronic kidney disease (CKD) is a common clinical feature of MM. In fact, despite aggressive treatment, progression to CKD Stage 5D occurs in ≤65% of patients with cast nephropathy within 3 months of diagnosis.17 Patients with MM have a higher mortality rate than other dialysis patients,17,30 and the survival decreases with more advanced myeloma.17 In addition, MM treatment-related morbidity and mortality are higher in CKD patients.31
Both haemodialysis and peritoneal dialysis are equally effective in patients with MM, with a comparable rate of treatment-related complications.17,32,33 Kidney transplantation may be considered in patients with MM who achieve sustained remission but remain dialysis-dependent.8 However, the risks in this population include MM recurrence, infection, and monoclonal Ig-mediated graft dysfunction.34-37 Therefore, most centres require MM treatment-free remission for at least 3–5 years for a patient to be eligible for kidney transplant.8,36
Light Chain Amyloidosis
AL amyloidosis is a systemic disease caused by the extracellular deposition of fibrils.16 It is most typically associated with lambda light chains.17 It is the most frequent glomerular lesion in patients with MM, found in ≤30% of cases.17,38 Approximately 50% have RI at presentation, >70% have proteinuria, and almost 30% present with nephrotic syndrome.38 Proteinuria is usually mild in patients with vascular limited AL amyloidosis.38,39 Other systemic symptoms include congestive heart failure, orthostatic hypotension, peripheral neuropathy, diarrhoea, macroglossia, and bleeding diathesis.38,39
On light microscopy, amyloid appears as eosinophilic, acellular, and weakly periodic acid-Schiff positive. It is distinguished from other hyaline deposits by their affinity for Congo red dye and birefringence under polarised light.16,17 After identification of amyloid and further characterisation and typing of the amyloidogenic component, it is essential to determine the target of therapy. On immunofluorescence, the deposits should stain for a single light chain in AL amyloidosis.38,39 Electron microscopy reveals randomly arranged fibrils with a diameter of 7.5–10 nm.16,17 The most common kidney compartment affected by amyloid deposits is the vessel walls, but deposits can be found in the mesangium and tubular interstitium.16
Progression to CKD Stage 5D occurs in one-third of patients,39 the median time from diagnosis to initiation of dialysis is ˜15 months, and median survival on dialysis ranges from 8–22 months.16,39 The median survival time in AL amyloidosis patients with nephrotic syndrome is 16 months.39 Lambda AL amyloidosis, RI, and cardiac involvement are associated with worse prognosis.17,39 In dialysis AL-amyloid patients, the primary cause of death is cardiac amyloidosis.16,38,39 Recent therapeutic advances for MM have increased haematologic response and improved median survival.40-43 Renal response, defined as a 50% decrease in proteinuria in the absence of a 25% increase in serum creatinine or a 25% decrease in glomerular filtration rate, is associated with haematologic response and is a marker of overall survival.44,45 Due to the poor prognosis of Stage 5D patients with AL amyloidosis, kidney transplant is rarely an option; however, it can be considered in patients without significant cardiac involvement, in combination with successful ASCT.37
Monoclonal Immunoglobulin Deposition Disease
MIDD occurs in 25% of MM patients.17,46 It includes light-chain deposition disease (LCDD), light heavy-chain deposition disease, and heavy-chain deposition disease, the most frequent being LCDD, which accounts for >70% of cases.16,46,47 In LCDD, kappa light chains are more frequent than lambda light chains.46,47
RI and proteinuria are the more common renal manifestations, typically rapidly progressive renal insufficiency and nephrotic range proteinuria.47,48 Cardiac involvement, such as congestive heart failure, cardiomegaly, or arrhythmias, and liver involvement, namely hepatomegaly, portal hypertension, or hepatic insufficiency, are present in almost 80% of cases.47,48 Gastrointestinal or neurologic manifestations are present in <30% of patients.47,48
On light microscopy, two-thirds of patients present with nodular mesangial sclerosis.16,17 These nodules are periodic acid-Schiff and silver positive, and Congo red negative. Immunofluorescence stains the specific light, heavy chain, or entire Ig in a linear pattern diffusely along the glomerular and tubular basement membranes and along vessel walls. Electron microscopy reveals electron-dense deposits in the same localisation as in immunofluorescence.47,48 In ˜20% of MIDD cases, there is co-existence of MCN, and less frequently of amyloid deposits.48
Patient survival and treatment response appears to be substantially better in LCDD than in systemic AL amyloidosis, with median patient survival reported as high as 14 years in some cohorts.46,48,49 Renal prognosis is poor, with most patients with MIDD progressing to CKD Stage 5D.48,49 Age and serum creatinine at presentation are predictors of CKD progression, and age and presence of extra-renal manifestations are also predictors of mortality.49
Early diagnosis and treatment is critical and may improve renal outcomes, as clinical remissions of LCDD with resolution of nodular glomerulopathy have been reported.49,50 Survival of MIDD patients on dialysis is variable, from 7–48 months.47,48 Due to the poor prognosis of MIDD, kidney transplant is not generally considered, and should be reserved for patients with a complete response to therapy, because the recurrence rate in patients not in complete response is nearly 80%.35
Less Common Renal Manifestations
Membranoproliferative glomerulonephritis is a less common MM-related kidney manifestation.51 It is an immune complex-mediated glomerulonephritis, characterised by subendothelial and mesangial deposition of immune complexes.51 Most patients present with RI, significant proteinuria, and hypertension.51 The prognosis is poor, with most patients progressing to CKD Stage 5D, and with frequent recurrence of the disease after kidney transplant.51
Fanconi syndrome is a rare disease, characterised by global proximal tubular dysfunction that results from crystalline deposition from a monoclonal light chain, most commonly kappa light chain.16,52 Most patients present with proteinuria, RI, and electrolyte abnormalities in the serum including hypouricaemia, hypophosphataemia, and hypokalaemia, and urine, including aminoaciduria, glycosuria, and phosphaturia.52 Rarely, distal tubular dysfunction occurs in association with proximal tubular dysfunction, presenting with distal renal tubular acidosis and nephrogenic diabetes insipidus.52 The renal prognosis is variable in these patients.53
Immunotactoid glomerulonephritis is a rare glomerular disease characterised by the deposition of fibrils in the glomerulus, distinguished from amyloidosis, as its fibrils are larger and do not stain with Congo red dye.54 It is often associated with a monoclonal gammopathy, and in 12.5% of these cases with MM. The typical presentation is heavy proteinuria, haematuria, and mild RI.54 Renal failure due to myeloma infiltration has also been documented, but only in 10–31% of autopsy series.55,56
CONCLUSION
MM-associated renal failure is associated with significant morbidity and mortality. Therefore, it is important to promptly diagnose and initiate adequate therapy. MCN is the most common cause of renal dysfunction in MM. The recent introduction of new chemotherapy agents and ASCT, and the development of novel techniques of light chain removal, have been associated with improved renal and patient outcomes in MM patients. Nevertheless, dialysis-dependent patients with MM have higher mortality than other dialysis patients, and may be considered for kidney transplantation only if sustained remission has been achieved and sustained for 3 years, bearing in mind the risk of disease recurrence.






