
Abstract
This review presents the recent advances in our understanding of the cellular pathogenesis of T cell non-Hodgkin lymphoma (NHL) and the potential of clinically targeted therapies. Patients with T cell NHL continue to face a limited prognosis, with the large majority experiencing a relapsed/refractory disease course and succumbing to their disease. Recent significant advances in our understanding of lymphomagenesis have not only revealed the complexity of T cell NHL but also helped to identify the cellular structures and pathways required for tumour proliferation, immune evasion, and therapy resistance. The NFκB pathway plays a critical role in T cell lymphoma through complex interactions with cell surface receptors and ligands, the proteasome, and crosstalk with ancillary pathways, such as the PI3K/Akt/mTOR cascade, which are also involved in chemokine and cytokine-mediated cellular signalling and growth. There is now also growing evidence for recurrent mutations involving the JAK/STAT pathway in a number of T cell lymphoma subtypes. Preclinical studies have highlighted the importance of novel cell surface proteins, downstream pathways, proteasome activation of NFκB, nuclear transport proteins, folate metabolism, epigenetic regulators, and cell of origin derivation. These advances represent a new era in T cell NHL therapy development. Although the optimal chemoimmunotherapy combination for first-line and salvage therapy is yet to be defined, the future paradigm is clearly shifting towards a biology-driven approach, which will hopefully yield improved outcomes for all patients with T cell lymphoma.
INTRODUCTION
The rapid evolution of laboratory technologies over the last decade has enhanced our ability to understand the intricate pathways involved in lymphoma biology and therapy resistance, heralding an era of novel, targeted, non-chemotherapy-based approaches to treatment.1 However, the outcome of most peripheral T cell lymphoma (PTCL) subtypes remains poor, highlighting the limitations of traditional chemotherapy and the importance of a biology-driven paradigm.2 This article presents an update on recent advances in T cell lymphoma biology by examining the current evidence for pathways implicated in disease and commenting on their therapeutic potential (Figure 1).
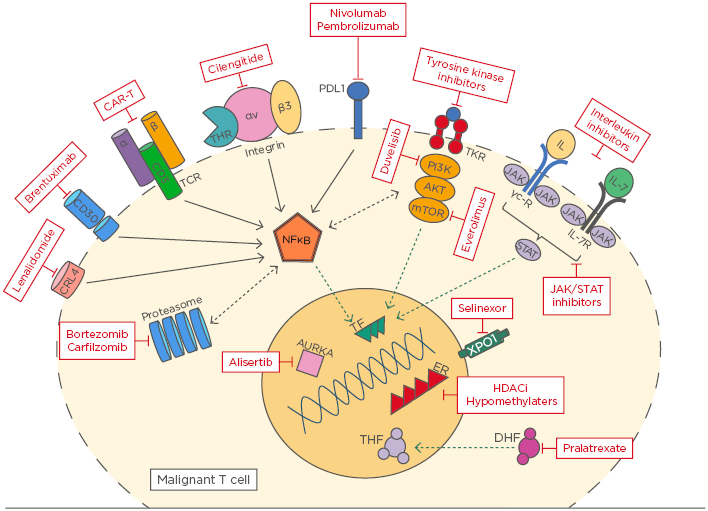
Figure 1: An overview of novel targets in T cell lymphoma.
AURKA: Aurora A kinase; CAR-T: chimeric antigen receptor T cell; DHF: dihydrofolate; ER: enhancer; HDACi: histone deacetylase inhibitor; TCR: T cell receptor; TKR: tyrosine kinase receptor; THF: tetrahydrofolate; TF: transcription factor.
THE NFκB PATHWAY
A common characteristic of various lymphoma and leukaemia subtypes is constitutive expression of NFκB, a master regulator of the inflammatory response.3,4 In the canonical (classical) signalling pathway, NFκB release is mediated by the activation of proinflammatory cell surface receptors (TNF receptor, IL receptor, Toll-like receptor, T cell receptor [TCR], epidermal growth factor receptor), activating the IκB kinase complex.4 In the non-canonical (alternative) signalling pathway, NFκB activation is triggered by signalling from CD40L, lymphotoxin receptor, and B cell activating factor receptor.5 Once activated, NFκB directly binds to DNA, propagating a number of pro-oncogenic changes. Its role in lymphomagenesis can be broadly separated into three categories: proinflammatory (upregulation of cytokines, such as IL-6, IL-8, and TNF-α, and chemokines, such as CXCL2), antiapoptotic, and induction of mitogenic proteins (e.g., c-Myc).4,6,7
As demonstrated by Wang et al.,8 tyrosine kinase interaction with the TCR signalosome (TCR proteins acting as a network hub, orchestrating interactions to control signalling)9 leads to activation of the NFκB pathway and production of specific transcription factors required for T cell lymphoma proliferation, immune evasion, and therapy resistance. Furthermore, NFκB activation has been shown to induce programmed cell death protein-1 (PD-1) expression and histone modification in T cells, macrophages, and B cells, leading to T cell exhaustion.8 With recent advances in small molecule therapy, it is now possible to target components and ancillary pathways involved in NFκB activation.10
CELL SURFACE TARGETS
CD30
The discovery of immunohistochemistry and the understanding that a proportion of T cell lymphomas express CD30 have led to the successful development of anti-CD30 as a therapeutic strategy, especially for anaplastic large cell lymphoma (ALCL), in which CD30 stimulation is known to upregulate NFκB activity.11 Brentuximab vedotin (BV) is an antibody–drug conjugate that targets CD30 in which the conjugated agent, monomethyl auristatin E, is a potent anti-tubulin toxin. BV has shown profound anti-tumour activity as a single agent in relapsed ALCL. Among 58 patients with relapsed/refractory ALCL treated with single agent BV, an overall response rate (ORR) of 86% and complete response (CR) rate of 57% was observed,12 which translated to a 5-year overall survival of 60%, an unprecedented survival rate in this patient group.13 Efforts are now being made to use BV in the upfront setting. Twenty-six patients with CD30-expressing T cell lymphoma (16 of whom had ALCL) were treated with cyclophosphamide, doxorubicin, and prednisolone in combination with BV. The ORR was 100%, with a 92% CR rate. The estimated 5-year progression-free survival (PFS) and overall survival were 52% and 80%, respectively, which compares favourably with historical data.14 The results of a subsequent Phase III, randomised, double-blind, placebo-controlled trial, ECHELON-2,15 comparing cyclophosphamide, doxorubicin, and prednisolone in combination with BV with standard cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) chemotherapy, are eagerly awaited.
TCRB1 and TCRB2
More recently, the ability to discriminate between normal and abnormal T cells through analysis of the TCR β-chain has been explored. There are two genes involved in the TCR β-chain constant region (TCRB1 and TCRB2), which are expressed in a mutually exclusive manner. Hence, a normal population of T cells will comprise a mixture of TCRB1+ and TCRB2+ cells.16,17 In their landmark paper, Maciocia et al.18 demonstrated tumour cell clonality, based on TCRB1+/- status, in peripheral blood, marrow, and tissue samples from patients with a range of T cell non-Hodgkin lymphoma (NHL) subtypes. Using a mouse model of T cell NHL, the researchers demonstrated the efficacy of targeting TCRB1-expressing malignant T cell clones with anti-TCRB1 chimeric antigen receptor-expressing T cells.18 These findings carry significant implications not only for therapy but also for diagnosis and disease monitoring.
Cell Surface Receptors
Cell surface receptors also provide a means for malignant T cell survival through interaction with external stimuli within the pathological niche. The transmembrane receptor integrin αvβ3, expressed on malignant cells, plays a pivotal role in this interaction. Thyroid hormones are known to exert their action through simultaneous binding of nuclear receptors and integrin αvβ3.19,20 Cayrol et al.21 demonstrated that thyroid hormones at physiologic levels can stimulate murine T cell lymphoma via intracellular pathways, ultimately leading to activation of NFκB and angiogenesis promotion. Furthermore, using a PTCL not otherwise specified (PTCL-NOS) xenograft model, Cayrol et al.21 demonstrated the therapeutic efficacy of cilengitide (a clinically available integrin αvβ3 inhibitor). Mice treated with cilengitide had a statistically significant reduction in tumour size, associated with decreased NFκB pathway activation and increased apoptosis.21
IL receptors also play an important role in the activation of the NFκB pathway. Recent evidence has identified a pathogenic role for IL-7 and IL-7R in T cell lymphomas.22-25 Using mouse models, Yasunaga et al.26 demonstrated that increased IL-7R signalling promoted tumour growth and steroid resistance in lymphoid malignancies. Conversely, inhibition of IL-7R signalling using an antibody–drug conjugate could effectively reduce tumour size, limit secondary lymph node infiltration, and potentially overcome steroid resistance.26
Programmed Cell Death Protein-1/Programmed Death-Ligand 1
As previously noted, NFκB pathway activation induces PD-1/programmed death ligand-1 (PD-L1) expression in a number of different cell types, including T cells and macrophages. Histopathological studies have confirmed increased PD-1/PD-L1 expression in a number of T cell NHL, especially in nodal and extranodal aggressive phenotypes.27-29 Thus, PD-1/PD-L1 blockade has been the focus of a number of early-phase studies in lymphoma.
Nivolumab, a fully human IgG4 monoclonal antibody (mAb) against PD-1, was noted to induce an ORR of 15% and 40% in mycosis fungoides (MF) and PTCL NOS, respectively, at a median follow-up of 67 weeks.30 However, it must be noted that the number of participants in this early-phase study was limited and no cases of CR were observed in the T cell NHL cohorts.30 Nevertheless, given the signal of favourable activity combined with an acceptable safety profile, further studies are underway exploring the potential for combining PD-1 inhibition with other agents.
Ansell et al.31 recently reported preliminary findings of a Phase I study of nivolumab and ipilimumab combination therapy, a human mAb targeting CTLA-4 (involved in the non-canonical NFκB pathway), in heavily pretreated NHL. Of the 11 T cell NHL subjects (7 cutaneous T cell lymphomas [CTCL] and 4 PTCL-NOS), only 1 (9%) achieved a partial response, 4 (36%) had stable disease, and no cases of CR were observed. The median overall survival in the T cell NHL cohort was 13.2 months, with a median progression-free survival of 2 months. Despite the limited number of participants, these results were favourable when compared to the B cell NHL cohort and similar to those noted in prior studies of nivolumab monotherapy.31
Chronic Epstein–Barr virus infection is known to induce PD-1/PD-L1 expression. Given its association with extranodal natural killer/T cell lymphoma (ENKTL), there has been increasing interest in its role in this subtype of T cell NHL. Retrospective studies have identified PD-L1 expression as a potential marker of favourable disease control, with improved OS and PFS noted in both advanced and nasal-type lymphomas.32,33 Although combination chemotherapy is still the preferred first-line treatment,34-37 novel therapies are being studied in the relapsed/refractory setting. In a recently published study, Kwong et al.38 treated seven ENKTL patients with relapsed/refractory disease following exposure to L-asparaginase-containing regimens. Patients received single agent pembrolizumab (anti-PD-1 mAb) at a fixed dose of 2 mg/kg at 3-weekly intervals (with the exception of one patient who was dosed at 2-weekly intervals). After a median of seven cycles of therapy and a median follow-up of 6 months, all patients demonstrated a response, with five (71%) meeting the criteria for CR and strength of PD-1 expression correlating with disease response.38
PI3K/AKT/mTOR PATHWAY INHIBITION
Although not directly linked to NFκB regulation, there is considerable cross-talk between the PI3K pathway and canonical activation of NFκB via AKT.39 Direct suppression of either pathway results in a reciprocal reduction in activity of the other.40 Various B cell lymphoma models have also demonstrated simultaneous activation of both pathways contributing to lymphomagenesis.41,42 In this regard, a number of recent studies have identified the sensitivity of B and T cell lineage leukaemia and lymphoma cells to autoimmunity checkpoint activation (to avoid clonal deletion by autoreactive B and T cell receptors), via upregulation of numerous pathways, including PI3K activation.43-45
Given that in vitro PI3K-δ/γ subunit inhibition can directly supress T cell lymphoma growth and proliferation, Horwitz et al.46 studied the activity of duvelisib (a novel PI3K-δ/γ) in a Phase I open-label trial in patients with relapsed/refractory PTCL-NOS (n=16) and CTCL (n=19), the majority of the latter group being histone deacetylase inhibitor-resistant. The researchers observed an ORR of 50% and 31.6% for PTCL-NOS and CTCL, respectively, with three patients achieving a CR. Changes in cytokine profile correlated with disease response, with an increase in soluble CD40L and IL-17α conferring a favourable outcome.46 Once again, despite limited participant numbers, preliminary data for PI3K inhibitors in T cell lymphoma is promising and the outcomes of a number of forthcoming clinical trials investigating novel combinations that include PI3K pathway inhibition are eagerly awaited.
The mTOR pathway is defined by the complex and inter-related activation of two distinct protein complexes, mTORC1 and mTORC2, which interact closely with the PI3K pathway and act upstream from and influence NFκB. Together, the interaction and activation of this protein complex leads to eIF4E and Akt activation, which promotes cell growth, survival, and proliferation in a number of malignancies, including T cell NHL.47-49
Preliminary studies, however, show that inhibition of mTORC1 alone leads to upregulation of Akt through disruption of its inhibitory influence on mTORC2.49 In vitro studies with everolimus (a first-in-class mTOR inhibitor) confirmed its inhibitory effect on mTORC1 and showed potential for efficacy in PTCL-NOS.50 This was confirmed in the clinical setting by Witzig et al.,51 following their study of 16 patients with relapsed T cell NHL (CTCL [n=7], PTCL-NOS [n=4], ALCL [n=2], ENKTL [n=1], angioimmunoblastic T cell lymphoma [AITL, n=1], and T cell acute lymphoblastic leukaemia/lymphoma [n=1]). The group observed a 44% (7/16) ORR and a median PFS of 4.1 months in response to a once daily oral everolimus dose of 10 mg. With a median duration of response of 8.5 months in responders, proof of concept was established.51 There are currently a number of early-phase trials underway testing the efficacy of second-generation mTOR inhibitors (which target both mTOR complexes) alone and in combination for relapsed/refractory T cell NHL. Furthermore, small molecules have been developed that can target both the mTOR and PI3K enzymes.52
PROTEASOME INHIBITION AND IMMUNOMODULATION
Despite the wide availability of a number of pharmacologic agents targeting the proteasome and considering its integral role in regulating the NFκB pathway (phosphorylation and ubiquitination of IκB), only a limited number of studies have assessed their role in T cell NHL. Zinzani et al.53 first reported on the efficacy of bortezomib (a first-generation proteasome inhibitor) in their 2007 Phase II study. They demonstrated a signal, predominantly in CTCL, with a 67% ORR that was sustained over 7–14 months.53 Subsequently, Ishida et al.54 demonstrated encouraging activity of lenalidomide monotherapy in adult T cell leukaemia/lymphoma (ATLL), which is prevalent in Japan, accounting for 25% of PTCL cases. In a Phase II study of 26 patients with relapsed ATLL, Ishida et al.54 observed an ORR of 42%, which met the study’s primary endpoint. This response was noted across all presentations of ATLL but the most encouraging response was noted in lymphomatous and unfavourable chronic presentations.54 More recently, through global transcriptome analysis, proteasome inhibition with ixazomib (a proteasome subunit beta type-5 inhibitor) significantly improved tumour response and overall survival in T cell NHL xenograft models via downregulation of Myc and checkpoint kinase-1.55
NUCLEAR TRANSPORTATION
The nuclear export receptor exportin 1 (XPO1) is a mediator of nuclear protein migration, including NFκB, and overexpressed in a number of haematological malignancies.56,57 Preclinical data demonstrated that inhibiting XPO1 led not only to an overall increase in cellular tumour suppressor proteins within malignant cells but isolated these proteins to the nucleus, promoting apoptosis and significantly prohibiting tumour cell growth and proliferation.56-58
Selinexor is a first-in-class oral selective inhibitor of nuclear export. In their study of 79 patients with relapsed/refractory NHL, Kuruvilla et al.59 observed an ORR of 31% (n=22), which included 4 cases of CR. The most prevalent safety concerns were Grade 3–4 thrombocytopenia and neutropenia, which occurred in 32% and 47% of patients, respectively. Tumour biopsies confirmed decreases in cell signalling pathways, reduced proliferation, and, most importantly, nuclear localisation of XPO1 cargos.59 Although T cell NHL patients were not included in this study, the pharmacodynamic results reported were very encouraging.59 However, the subsequent Australian Phase II study of single agent selinexor in relapsed/refractory T cell NHL60 was terminated early due to enrolment challenges (n=16 at time of study closure), with the results yet to be published. Despite this, the Singaporean National Cancer Centre has recently launched a Phase I trial of selinexor in combination with standard dose ifosfamide, carboplatin, and etoposide for relapsed/refractory PTCL.61 Furthermore, in vivo studies using eltanexor, a second-generation selective inhibitor of nuclear export, have shown early promise, with clinical trials forthcoming.62
THE COMMON GAMMA: JAK/STAT
The common gamma (γc) receptor-dependent cytokines (IL-2, IL-4, IL-7, IL-9, IL-15, IL-21) and their receptor targets play a critical role in T cell immunity. The receptors for these respective cytokines lack intrinsic kinase activity and, as such, their functionality is dependent on their association with JAK cytoplasmic tyrosine kinases. Cytokine binding to receptors leads to cross-JAK phosphorylation, phosphorylation of the intracellular cytokine receptor tail, and creates a docking site for STAT. Once activated, phosphorylated STAT translocates to the nucleus and acts as a transcription factor.63 Almost all forms of T cell lymphoma have now been associated with disorders involving activation of the γc/JAK/STAT system.64-71 It is now understood that activation of the γc/JAK/STAT system alone is not sufficient to cause abnormal T cell proliferation. For this to occur, the entire pathway, from cytokine receptor augmentation to STAT phosphorylation and nuclear transportation, must be activated.63 There is growing preclinical evidence for the efficacy of JAK/STAT inhibitors in T cell NHL72 and this will undoubtedly transfer into the early-phase clinical setting.
AURORA A KINASE INHIBITION
Aurora A kinase is a serine/threonine kinase that plays an integral role in cellular mitosis by localising to the centrosome and regulating chromatid segregation from prophase to metaphase. While expression is limited in normal tissue, overexpression has been identified in a number of malignancies, including subsets of T cell NHL.73,74 Subsequently, upon development of the selective oral aurora A kinase inhibitor, alisertib, a number of early-phase studies were performed.75-77
Initial results from two pivotal Phase II studies of relapsed/refractory lymphoma revealed an ORR of approximately 30%, with a promising signal in PTCL-NOS and transformed mycosis fungoides;75,76 however, these results have not translated into improved patient outcomes. As presented by O’Connor et al.,77 the interim analysis of the multicentre, randomised Phase II study of alisertib versus investigator choice for relapsed/refractory PTCL failed to meet significance and resulted in the study being prematurely terminated. Of the 238 patients randomised, the ORR of alisertib and investigator choice were 33% versus 43%, including a superior CR rate in favour of the latter.77 Despite these findings, the combination potential of alisertib remains to be explored. This result, however, underscores the importance of subjecting novel agents to randomised trials against standard of care before their adoption into routine practice.
EPIGENETIC DYSREGULATION
Gene transcription is not only dependent on a number of intracellular pathways but also relies on DNA interaction with the histone protein octamer, commonly known as the nucleosome. This interaction is largely controlled by post-translational modification of the histone protein, including acetylation and methylation.78 Direct methylation of cytosine bases within DNA is an additional mechanism of transcriptional control that is often linked to histone modification patterns and these processes together are termed epigenetic regulation. Mutations of enzymes involved in post-translational modifications lead to aberrant DNA methylation and promote oncogenesis. With respect to T cell NHL, studies have demonstrated recurring mutations in TET2, isocitrate dehydrogenase (IDH), and DNA methyltransferase 3 (DNMT3). Both TET2 and IDH mutations result in increased DNA methylation, thus establishing a role for demethylating agents and histone deacetylase inhibition in T cell NHL.2
Delarue et al.79 studied 19 patients mainly with relapsed/refractory PTCL (as first-line therapy for 2 patients) who were treated with the hypomethylating single agent 5-azacyctidine. Ten patients had a previous or concomitant diagnosis of myelodysplastic syndrome, mainly chronic myelomonocytic leukaemia. A 53% ORR was observed but 9 out of 12 patients (75%) with AITL responded.79 Of interest, eight patients with AITL who had Tet2 mutation status available had a mutation. A number of trials are ongoing using demethylating agents in combination.80
Promising early-phase data using first-generation histone deacetylase inhibitors (HDACi) led to the rapid approval of a number of agents for a variety of T cell NHL subtypes.81-86 However, single-agent activity is modest and has meant that while approved in the USA, European approval has not been forthcoming. In addition, randomised trials of single agent HDACi with standard of care have not been conducted, undermining confidence in their use. When faced with a modest activity signal as a single agent, focus has rightly shifted to combination therapies.
In their Phase Ib/II study of first-line romidepsin plus standard CHOP, Dupuis et al.87 demonstrated significant responses, albeit with associated myelotoxicity and potential cardiac toxicity. The ORR of 68% (51% CR, 17% partial response), PFS of 57%, overall survival of 76.5%, at a median follow-up of 17.5 months, came at a cost, with two-thirds of patients experiencing at least one serious adverse event.87 However, given the promising response and survival outcomes, the planned Phase III trial of romidepsin plus standard CHOP study88 was initiated.
The novel combination of HDACi and proteasome inhibitors also shows considerable promise. In their Phase II study of panabinostat plus bortezomib for relapse/refractory PTCL or NK/T cell NHL, Tan et al.89 reported an ORR of 43% (10 out of 23), with 22% (5 out of 23) of patients attaining a CR. The median time to response was 6 weeks. Myelotoxicity was once again identified as the major concern, with approximately two-thirds of patients experiencing Grade 3/4 haematotoxicity.89 The encouraging results of this study have led to second-generation combination therapies, with a study of romidepsin plus carfilzomib for relapsed/refractory PTCL currently recruiting in the UK.90 This study is also investigating the potential utility of HR23B protein expression as a predictive biomarker of response. HR23B was identified in a genome-wide loss-of-function screen to identify genes involved in the sensitivity of tumour cells to HDACi.91 The protein has an important role in shuttling ubiquitinated proteins to the proteasome.92 Retrospective studies have identified an association between HR23B expression and response of cutaneous T cell lymphoma.93 Prospective confirmation is required before its use as a predictive biomarker is established.
FOLATE METABOLISM
Neoplastic T cell proliferation depends on DNA and RNA synthesis, which require folate metabolism.94 Pralatrexate is an antineoplastic folate analogue that directly targets both cellular folate transport and metabolism through enzyme inhibition, disrupting DNA and RNA synthesis.95 Early in vitro studies demonstrated the cytotoxic activity of pralatrexate in a number of lymphoproliferative disorder cell lines and xenograft models.96-98
The pivotal PROPEL study,99 a Phase II single-arm, open-label study, enrolled 115 patients with relapsed or refractory PTCL. Of the 109 evaluable patients, the ORR was 29% (n=32), which included 11% CR (n=12) and 18% partial response (n=20). The median duration of response was 10 months, with a median PFS and OS of 3.5 and 14.5 months, respectively.99 The early durable responses observed led to the rapid approval of this agent in the USA for relapsed/refractory PTCL. Similar to with HDACi, no randomised trial was performed and a European license has not yet been granted.
The recent Phase I/II study of pralatrexate in Japanese patients supported the PROPEL data. Following an identical dosing regimen, the authors reported an ORR of 45% among the 20 evaluable Japanese patients.99,100 Although both studies demonstrated high rates of mucositis, this may be mitigated by concomitant leucovorin administration, thus enhancing the safety of this combination.101
The efficacy of pralatrexate in relapsed/refractory disease has led investigators to explore its combination potential. Shustov et al.102 recently presented preliminary data from their Phase I dose-escalation study of upfront pralatrexate 30 mg/m2 per day for 1–8 days, plus standard dose CHOP. Of the 27 evaluable patients, the researchers observed an investigator-assessed ORR and CR of 89% and 67%, respectively. The only treatment-related Grade >3 adverse events noted were neutropenia (n=4). The maximum tolerated dose of pralatrexate was not reached.102 Although preliminary, the relative safety of pralatrexate plus CHOP is reassuring and no doubt the efficacy of this and other similar novel combinations will be explored further with planned Phase II and III studies.
FOLLICULAR HELPER T CELL DERIVATION
A subset of CD4+ T cells, follicular helper T (Tfh) cells, play a critical role in physiologic immunity.103 Localised in lymphoid organs, Tfh cells have features of both central and effector memory T cells104 and, in comparison to normal B and T cells, Tfh express high levels of inducible costimulator (ICOS) and chemokine receptor 5. Through the influence of ICOS activity, Tfh cells undergo differentiation, with a high affinity for expression of BCL6, IL-24, IL-4, CXCL13, and PD1, in addition to ICOS and chemokine receptor 5.103,105-108 ICOS ligand activation is also closely linked with NFκB regulation.109 Thus, Tfh cells are a key effector cell at the interface between innate immunity and normal B and T cell maturation, with dysregulation leading to both autoimmune dysfunction and lymphomagenesis.
Gene expression studies of malignant T cells in AITL have established a striking similarity to Tfh cells, sufficiently supporting their role as the cell of origin in AITL. This is further supported by the biochemical and clinical features of AITL, including autoimmune dysregulation and enhanced B cell activation. Subsequent targeted sequencing studies have demonstrated recurrent mutations of TET2, DNMT3A, and IDH2, and loss of RHOA (coding for GTPase Rhoa) in AITL of Tfh cell origin.110-112 With this knowledge, Tfh cells have also now been defined as the cell of originin subsets of other T cell NHL, including PTCL-NOS and CTCL.
With respect to therapeutic implications, establishing Tfh cell derivation by pathologic and genetic analysis may select for tumour types that are sensitive to direct antibodies that target highly expressed antigens (e.g., ICOS, PD1, CXCR13). These tumours may also be susceptible to NFκB pathway (also regulated by ICOS ligand activation) targets, demethylating agents, and histone deacetylase inhibition. Further studies are required to establish the true impact of Tfh cell derivation in T cell NHL.
CONCLUSION
As it stands, the majority of patients presenting with T cell NHL will not successfully achieve a complete remission with current first-line standard of care chemotherapy, subsequently experiencing a relapsing/remitting course and eventually succumbing to their disease.113-116 Of the small proportion of patients who do achieve a favourable response, there is no consensus on consolidation approaches and those unsuitable for transplantation are likely to experience disease relapse.117-119 There is an urgent and unmet clinical need to improve the limited prognosis faced by these patients.
With a rapidly evolving understanding of tumour biology, we are gradually able to unravel the underlying biology of the various T cell NHL subtypes and exploit it to our advantage. One of the major hurdles to managing the disease is our inability to specifically target neoplastic T cells without diminishing the innate immunogenicity of normal T cells. As outlined, the medical community is now approaching an era in which we will not only be able to target the cells of origin, but we will be able to customise therapy based on identifying a patient’s unique immunophenotypic, histopathologic, and genetic features (Figure 1). Just as combination immunochemotherapy has dramatically changed the outcomes for B cell lymphoma, we now have the knowledge and the tools required to reshape the future of T cell NHL. Of those targets discussed, targeting cell surface molecules, such as CD30, with antibody–drug conjugates has changed standard of care for relapsed ALCL and may impact front- line treatment of CD30-positive tumours. To further impact survival, it is unlikely that a single target will suffice; rational combinations of targeted agents (with or without more conventional chemotherapy) in biomarker- defined populations represent the way forward in this heterogeneous disease.






