
Abstract
Ovarian cancer (OC) is a disease that almost invariably relapses even after optimal primary cytoreductive surgery and standard first-line platinum-based chemotherapy. After recurrence, progressions occur at shorter intervals in the natural history of the disease. However, the biologic and cellular events underlying recurrence and progression (maintenance phase) are yet to be completely understood. Ovarian adenocarcinoma, like any other tissue, after reduction of the cell population (cytoreduction) either by surgery, chemotherapy, radiotherapy, or targeted therapies induced cell-death, tends to its own renewal through cancer stem cells (CSC). CSC remain quiescent most of their lives and then ‘wake up’, generating a proliferative progeny that differentiates as they become different clones of daughter cells. What defines them is their ‘self-renewal’ potential, thus perpetuating the disease with higher tumour volume relapses in which CSC increase in number.
We propose a theory of how recurrence/relapse occurs in which CSC play a key role in the genesis of relapse. These self-renewing CSC can generate a proliferative progeny and this population is sensitive to chemotherapy, anti-angiogenic agents, and PARP inhibitors, which so far have only increased the disease/relapse free survival (‘maintenance phase’). In OC it seems we are not addressing the ‘root’ of recurrence/relapse. As with any theory, this is based on both proven facts and suggested hypotheses, which may serve as investigation drivers towards finally making a substantial improvement in OC management.
INTRODUCTION
Ovarian cancer (OC) is a disease of multiple relapses (Figure 1).1 After achieving a complete response with first-line platinum-based chemotherapy, >70% of the patients recur at 2 years and subsequently, progression is almost unavoidable; >70% show progression at 1 year.2-11 Many efforts have been made in order to improve these results: delivering chemotherapy intraperitoneally, in a dose dense schedule, or even adding a third agent to the standard carboplatin-paclitaxel backbone, which remains after >10 years. Antiangiogenic agents have also been incorporated after first-line treatment, but as with all the other attempts, only improvement in progression-free survival (PFS) with no impact on overall survival (OS) has been reached.
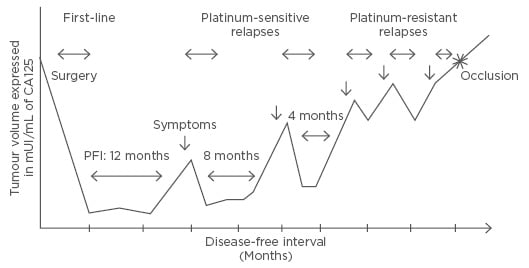
Figure 1: Natural history of ovarian cancer evolution.1
PFI: platinum-free interval.
The same concept applies for treatments after relapse (maintenance therapies): the use of antiangiogenic agents (bevacizumab, cediranib, trebananib)6,12-14 or PARP inhibitors, such as olaparib,15 only delays progression, with PFS becoming shorter (Figure 1).
The objective of this publication is to provide a theoretical view of OC relapses, focussing on their biological genesis, and to envision potential targets that may provide a substantial benefit in OC patient management. For this we performed a PubMed search, restricted to full-text articles, using the following search terms: ‘ovarian cancer’, ‘cancer stem cells’, ‘relapsed ovarian cancer’, and ‘ovarian cancer maintenance’.
BIOLOGY OF RELAPSE
As stated before, recurrence seems to be an unavoidable event in OC history since >70% recur even after being optimally debulked (R0) at first cytoreductive surgery and standard platinum-based chemotherapy. These recurrences/progressions can occur at varying intervals and have been classified according to platinum-free interval (PFI), the interval in months since the last platinum-dose, into the following categories:
- Platinum-refractory/resistant: PFI: <6 months
- Platinum-partially sensitive: PFI: 6–12 months
- Platinum-sensitive: PFI: >12 months
This classification was derived from retrospective data from Phase III randomised trials showing that efficacy of platinum-based chemotherapy was directly proportional to the PFI in terms of overall response rate (ORR), PFS, and OS.16,17 This distinction reflects that OC is an heterogeneous disease with different aggressive biology phenotypes, and also phenotypically, with mucinous and clear cells having a worse outcome than their more frequent and chemo-sensitive counterparts high-grade papillary serous and endometroid subtypes.18
Nowadays, we know that microscopic disease that survives first-line chemotherapy and second or third (at relapse) is responsible for recurrent disease or progressive disease, yet responds to the same chemotherapy agent (platinum) with ORR ≥70%, especially in BRCA mutated patients.19 In the homologous-recombination deficient (HRd) population (which is estimated to be 50% of the patients),20 the use of maintenance therapy with olaparib increased PFS by 9.4 months, but with no impact on OS.11 Coincident with these results, Phase III trials using antiangiogenic agents (bevacizumab, cediranib, trebananib) as maintenance therapy after platinum-sensitive or refractory relapse only prolonged ORR and PFS by targeting angiogenesis concurrent to and after chemotherapy.10,13,14,21,22 Although these different agents have proven to be effective in delaying progression, this still occurs at shorter intervals. Many unresolved issues remain about the biology of this disease diagnosed at relapse. Table 1 summarises some unanswered questions about relapse.

Table 1: Unsolved issues about cancer stem cells.
CSC: cancer stem cells; OC: ovarian cancer; ORR: overall response rate; PFS: progression-free survival.
THE CANCER STEM CELL THEORY
We know that tumours are heterogeneous not only in their histopathology, but for a certain type of tumours, like high-grade papillary serous, in the function of cells that comprise them, their proliferative capacity, as well as responsiveness to different agents (chemotherapy or antiangiogenics).
The CSC theory proposes that within a tumour there is a certain hierarchy with CSC being responsible for recapitulating the disease after cytoreduction generated by surgery, chemotherapy, or both. These CSC, which represent only a small portion of the tumour (<1%), a proportion that increases at relapse (self-renewal capacity), remain quiescent in the G0 phase of the cell cycle and therefore, not vulnerable to chemotherapy agents which damage DNA or microtubules in the S or M phase of the cell cycle. They are tumourigenic (able to create tumour foci when injected in immunosuppressed mice),23-26 clonogenic (able to create a proliferating, differentiating progeny),27,28 and pluripotent (not only able to generate tumour cells but microenvironmental cells as well).29 It is their proliferating progeny that are vulnerable to chemotherapy agents, especially because OC cells are deficient in repairing chemotherapy- damaged DNA.30,31
Thus, according to this theory, OC may be regarded as an aberrant tissue that arises from CSC, which in turn may have arisen from normal stem cells (SCs) after a mutation that generated this self-renewal phenotype, or from a mutation in a normal cell that allowed it to dedifferentiate into a CSC or early progenitor.32 Hence, they create a ‘back-up’ population that is able to recapitulate the disease. This self-renewal capacity is what defines this progeny with a ‘SC-like phenotype’.
CSC are a subpopulation of cells within the tumour that have different markers of chemo and radio-resistance (CD133, aldehyde dehydrogenase, CD44) as well as CD24 (related to tumour-initiating capacity).31 They have the capacity to repopulate foci of tumour at sites of primary undebulked disease after achieving complete response due to chemotherapy, but can also metastasise early to distant microscopic foci, generating OC relapse/recurrences.20,33 Changes in the microenvironment or any other ‘driver-event’ or ‘transforming-event’ (endocrine, paracrine, or autocrine factors) wake them up from that quiescent state and allow them to proliferate asymmetrically: one daughter-cell repopulates the CSC pool and the other starts to proliferate, generating a progeny with different pathways of differentiation (some cells can differentiate to endothelial cells depending on the stimulus that the microenvironment provides) or even dedifferentiate to a more ‘SC’ phenotype.34 Figure 2 shows CSC differentiation and tumour-cell hierarchy in OC based on the current literature.
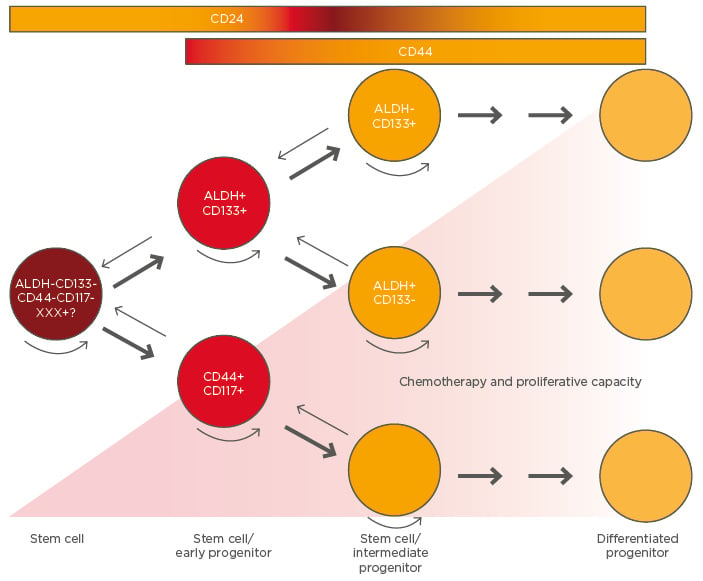
Figure 2: Hierarchy in ovarian cancer and cancer stem cell differentiation.
Rounded arrows represent self-renewal capacity. Small arrows indicate the unproven potential for dedifferentiation.
ALDH: aldehyde dehydrogenase.
Adapted from Burgos-Ojeda et al.31
CSC act as an insurance population and are responsible for relapses and progressions in OC in which CSC population is higher.35 There is clinical evidence to support this CSC hypothesis in OC, and it has also been extensively studied in other tumour models, such as breast, colon, and leukaemia.36-40
CLINICAL EVIDENCE TO SUPPORT THE CANCER STEM CELL THEORY IN OVARIAN CANCER
Although not extensively studied in terms of their impact on OC prognosis, some evidence suggests that CSC correlate with a high volume of disease at recurrence and a worse prognosis.31 To date, the only therapeutic manoeuvre with a positive impact on OS is macroscopically removing all foci of tumour (R0)41 harbouring CSC, in other words rendering the patients optimally debulked, or R0, at the time of primary cytoreductive surgery. Nevertheless, clinical evidence suggests that microscopic foci of disease also harbour CSC generating recurrent/progressive disease.33
High Response Rates at Relapse with Same Chemotherapy Regimens
As stated earlier, the longer the disease-free survival (DFS)/PFS, the better the results with platinum based chemotherapy. If recurrence/relapses were generated by surviving chemo-resistant clones, why does the platinum-sensitive relapse have an ORR >70% with the same chemotherapy combination?42
The high ORR are due to the effect that chemotherapy has on a proliferating progeny with a frequent mutation in p53 (a critical checkpoint for cell-cycle progression) and a HRd phenotype in 50% of the patients.43 p53, BRCA1, and BRCA2 mutations (the hallmark of HRd) are described as ‘early driver-events’ in OC carcinogenesis. Is the platinum-refractory/resistant disease richer in CSC or is chemo-resistant chemotherapy an early progenitor?
Trials with Antiangiogenic Agents as Maintenance Therapies
Two pivotal Phase III trials (GOG 218 and ICON 7)6-12 with bevacizumab and one with pazopanib (AGO-OVA),44 after first-line chemotherapy, demonstrated a benefit in DFS for patients treated with bevacizumab, especially in high-risk groups: suboptimally debulked and Stage IV.
In the relapse setting, these agents have been evaluated as well: the OCEANS trial for platinum-sensitive relapse and AURELIA for platinum-resistant relapse. In both, the addition of bevacizumab to chemotherapy not only improved ORR but also PFS. In the ‘platinum-resistant relapse’, the patients who benefited the most were those who presented with ascites, in whom bevacizumab improved global and disease-specific quality of life scores.10,21,22 ICON 6 evaluated cediranib,13 a vascular endothelial growth factor (VEGF) tyrosine-kinase inhibitor, as maintenance after platinum-sensitive relapse. The experimental arm that received cediranib as maintenance had a significant improvement in PFS (hazard ratio [HR]: 0.56; 95% confidence interval [CI]: 0.44–0.72).
It is well known that the tumoural microenvironment changes after chemotherapy, with haemorrhagic necrosis in sites of chemotherapy-induced response. This implies an improvement in hypoxic conditions. This microenvironmental improvement may be a ‘driver event’ for CSC to abandon their quiescent state and regenerate the tumour, because conditions are now more favourable.45 However, clinical evidence shows that targeting the VEGF pathway, either through bevacizumab or cediranib, does not create a situation of chronic hypoxia for CSC to remain quiescent and the proliferating progeny finds alternative ways of angiogenesis or survival under these hypoxic conditions,46,47 as patients who relapse after getting bevacizumab first-line do not derive more benefit by adding it to chemotherapy at relapse.48
PARP Inhibitors in Maintenance Trials
PARP inhibitors are the first types of agents approved in OC based on a predictive biomarker for response (BRCA1 and BRCA2 mutations).
This subgroup of OC patients, which represents 14% of cases considering germline-mutation positive (gBRCAm) but ≤20% if somatic-mutation (sBRCAm) patients are added,43 have the HRd phenotype, meaning that the most faithful, error-free, DNA damage-repair mechanism for DNA double strand damage is inefficient. This kind of damage is caused by carboplatin: double strand DNA-adducts, which can only be efficiently repaired by homologous recombination. Many factors interact with this mechanism, but BRCA1 and BRCA2 play a key role. The mutations of the genes that encode for these nuclear proteins are the most prevalent mutations leading to this HRd phenotype.49 Other genes involved in homologous recombination are less frequently mutated and lead to a ‘BRCAness or BRCA-like’ phenotype (HRd phenotype). These types of tumours are less capable of repairing the damage chemotherapy induces in their DNA, and, consequently, are more chemo-sensitive. In fact, BRCAm OC has a better prognosis than BRCA wild-type (BRCAwt).50
PARP inhibitors create a situation in which OC cells are forced to use this HRd mechanism. By trapping PARP, a key factor in one of the mechanisms for repairing single-strand DNA breaks, like the one reactive species of oxygen created in the hypoxic tumour microenvironment, they create a situation of ‘synthetic lethality’: single-strand DNA breaks become double-strand breaks as the replication fork gets stalled inside the double helix by the PARP inhibitors. These agents take advantage of this cell deficiency and create a genomic instability state, making the cell utilise an alternative method of double-strand DNA damage repair; non-homologous end joining in which essentially, the injured DNA is excised and the proximal and distant ends joined, producing extensive loss of genetic material, which is critical for cell survival.51,52
Study 19 is a Phase II trial that evaluated the activity of olaparib, the most extensively studied PARP inhibitor in OC, as maintenance therapy after platinum-sensitive relapse (a surrogate of HRd). They demonstrated a benefit in PFS for the global population (HR: 0.65; 95% CI: 0.25–0.49), but the difference was greater among BRCA1 and BRCA2 mutated patients (germline or somatic) who received maintenance with olaparib: (HR: 0.18; 95% CI: 0.11–0.31). This agent is the first target agent to show such a PFS benefit with a low toxicity profile (12% patients on treatment at 5 years), given orally. Nevertheless, no benefit in OS was demonstrated for the olaparib treated patients, not even the BRCAm.11
This suggests that even though we can target a defective mechanism of utmost importance for cell-survival (DNA repair), the HRd proliferative progeny either develops mechanisms of resistance or alternative repair mechanisms to survive despite these mutations.
Combination of Antiangiogenic Agents and PARP Inhibitors
Liu et al.53 presented the data of a Phase II trial in which patients with platinum-sensitive relapse were randomised either to olaparib or a combination of olaparib plus cediranib. This trial showed that the combination favoured BRCAwt population in terms of PFS over BRCAm patients. The reason behind this finding is the concept of ‘contextual synthetic lethality’ in which cediranib-induced hypoxia downregulates replication, transcription, and transduction of factors involved in homologous recombination, making the BRCAwt (homologous recombination ‘efficient’) behave as HRd.54 This combination is being evaluated against a platinum-based combination and olaparib in platinum-sensitive relapse as a chemotherapy-free regimen, and the efficacy will very likely be the same. However, this combination did not significantly improve OS, even targeting two mechanisms that are part of the biology underlying relapse/progression (angiogenesis and DNA damage repair response). Therefore, unless we target the real source of relapse: CSC, OC will still be considered a disease of multiple relapses.
CANCER STEM CELLS AS A TARGET
One of the most important hurdles to clear in targeting CSC is the lack of knowledge of their biology: why or what makes them remain in this quiescent state that renders them less vulnerable to chemotherapy or DNA damaging therapies. It seems that hypoxic conditions lead to the expression of a ‘SC-like’ phenotype as a way of the tumour ensuring its perpetuity. What are the ‘driver-events’ that lead them to enter in cell-cycle, apart from improvement in tumour micro environment? Is it a paracrine factor or even an endocrine factor whose systemic concentrations are low when tumour-burden is low after chemotherapy or surgery induced cytoreduction?
It has been demonstrated that the Hedgehog, Notch, and Wnt pathways are key signalling pathways for CSC differentiation and apoptosis.55 Through these mechanisms CSC develop the whole differentiation ‘programme’ (plasticity). Other histological subtypes, which account for less than a third of OCs, probably are driven by a different type of differentiation either by genetic or environmental factors. The rare types: clear cell, mucinous, endometroid, and low-grade adenocarcinoma, have their own driver mutations (PTEN, PI3K/AKT, ARID1A, Her2/neu, BRAF, KRAS)56-58 which explain some of their biological behaviours, and also possess new targets for the maintenance phase after chemotherapy (in which no chemotherapy regimen other than standard carboplatin/paclitaxel has been demonstrated to be better in first-line therapy, regardless of the subtype). In this range of subtypes, low-malignant potential tumours (borderline tumours) might represent the most differentiated form. In fact, Emmanuel et al.59 demonstrated in 14 micro-dissected specimens with borderline and invasive tumours coexisting in the same sample that chromosome copy number aberrations were remarkably conserved in the invasive and borderline components, suggesting that the latter could be the ‘fully-expressed differentiation programme version’ of the invasive form. Is there a way to promote this differentiation of the CSC generated progeny?
CSC express markers like OCT4A, NANOG, and SOX2. These proteins are fundamental for differentiation and survival. T cell reactivity against OCT4A has been demonstrated, making it targetable for vaccination and inducing an anti-CSC memory immune response. Ongoing trials are evaluating dendritic cell vaccination against these CSC antigens.60
CONCLUSIONS
Although efforts have been made to improve the results of first-line chemotherapy, as well as treatment at relapse, only improvements in DFS/ PFS have been accomplished without a dramatic impact on OS. Over the last 30 years not much has changed in OS at 5 years with <30% of patients remaining alive.
The biology and the underlying cellular and molecular events that take place during the maintenance phase (variable periods between relapses) are far from being completely understood. Targeting two of the known mechanisms that lead to progression, angiogenesis, and DNA damage repair mechanisms, have shown similar results (increased PFS, no benefit in OS). Unless the root of progression: CSCs or tumour initiating cells are targeted, OC will still be a chronic disease with multiple relapses.






