Meeting Summary
Myasthenia gravis (MG) is an autoimmune disease caused by autoantibodies targeting proteins on the neuromuscular junction. Around 85% of patients have antibodies against the muscle acetylcholine receptor (AChR), while up to 10% have antibodies against the muscle-specific kinase (MuSK). Rarer forms of the condition are low-density lipoprotein receptor-related protein 4 (LRP4) positive MG, and seronegative MG, in which no AChR, MuSK, or LRP4 autoantibodies are detected. MG, which can be ocular or generalised, is characterised by muscle weakness, which can be severe and debilitating.
Despite advances in treatment, a significant proportion of patients living with generalised MG (gMG) continue to experience symptoms and poor quality of life (QoL). During this symposium, John Vissing, Professor of Neurology at the University of Copenhagen, and Director of the Copenhagen Neuromuscular Centre, Denmark; Heinz Wiendl, Professor of Neurology at the University of Münster, Germany; and Kristl Claeys, Professor of Neurology at the University of Leuven, Belgium, discussed the current challenges and future potential of MG treatments. They emphasised the need for patient-centred evaluations, discussed the pathophysiology, and highlighted the challenges of current immune therapies. They also explained how new generations of targeted immune therapies, such as neonatal Fc receptor (FcRn) inhibitors, could help tackle this area of unmet need by potentially ameliorating disease manifestations.
Introduction
MG is a rare immunoglobulin G (IgG) autoantibody-mediated disease, characterised by fluctuating weakness of the voluntary muscles.1 The weakness, which tends to worsen with activity as the day progresses,1 is caused by the pathogenic IgG-mediated disruption of cholinergic transmission at the neuromuscular junction.1,2 The majority of people with MG, around 85%, are AChR+, and up to 10% have antibodies against MuSK.3 Rarer forms of the condition are seronegative MG, in which no AChR, MuSK, or LRP4 autoantibodies are detected, and LRP4+.3
In ocular MG, the weakness affects the extraocular muscles, manifesting as diplopia and palpebral ptosis.1 gMG can also affect the bulbar muscles (causing difficulties with speaking, swallowing, and chewing), the extremities (impeding gait and everyday activities), or axial muscles (causing weakness in the back and neck, and leading to painful spasms).1 gMG treatments include cholinesterase inhibitors, immunosuppressants, and targeted immunotherapies; yet, symptom persistence, exacerbations, and side effects are common with current therapies.2
Optimising Patient Outcomes and Satisfaction
While the past few decades may have seen a drastic reduction in gMG mortality,4 many patients still experience symptoms and poor QoL.5,6 “We have come a long way,” said Vissing, explaining that in the decade following diagnosis, many patients become asymptomatic, “but we still have quite a big group of patients, 15–20%, who have significant symptoms at two years.”7 Illustrating the impact this can have on people’s lives, Vissing shared a video of a 41-year-old female talking about living with gMG. Her disease manifestations, the patient said, included difficulty breathing, chewing, swallowing, and speaking, as well as cognitive challenges, which can vary from “day to day, week to week, and month to month.” “I might have reasonably well-controlled symptoms in the morning, but by the evening I may not be able to get up and down the stairs,” she said, adding that when her symptoms were under control, she lived “what looks like a normal life on the outside.” “But I miss out on a lot of things because I am very aware of not overextending or asking too much of my body and causing an exacerbation. […] It can be a very lonely disease […] because you have to continually cancel plans […] and miss out on the things that give life texture and colour,” the patient said.
Patient-Centred Evaluation
Currently, there are limited data on the time it takes to reach an acceptable state, or remission, in MG, and most studies assessing long-term outcomes are rated by clinicians rather than patients, said Vissing. Minimal symptom expression (MSE), defined as an MG Activities of Daily Living (MG-ADL) total score of 0–1,8 could be a patient-centred way to measure disease impact. In a study of 85 patients with AChR+ refractory gMG treated with immunotherapy, 55.8% had reached MSE by Year 1, and 60.3% at Year 2.9 “The amount of time to achieve MSE in patients undergoing immunotherapy was very long, and not all patients managed to achieve it,” said Vissing, adding that another study had shown patients with a high disease burden (MG‐ADL score ≥6) “rarely achieved” MSE after 1 year of treatment.10
The Patient Acceptable Symptom State (PASS), which defines a threshold of the individual’s satisfaction with their MG status, is another patient-centred method of assessing symptom burden.11 It consists of a single, dichotomous question, i.e.: “Considering all the ways you are affected by myasthenia gravis, if you had to stay in your current state for the next months, would you say that your current disease status is satisfactory?”11 Response options are “yes” or “no”.11 Studies using PASS have shown that patient dissatisfaction with disease state is common. In one study of 100 patients living with gMG, one-third reported a negative PASS status, meaning they were dissatisfied with their current symptom state.12 Increasing MG symptoms, fatigue, depression, low MG-related QoL, and shorter disease duration were all associated with a negative PASS status.12 In another study of 86 patients with MG, the median time to reach a positive PASS status after a post-diagnosis negative PASS status was 15 months (95% CI: 11–18).11 Of the 67 patients with MG who achieved PASS (“yes”), 61 (91%) achieved it by 25 months post-diagnosis (Figure 1).11
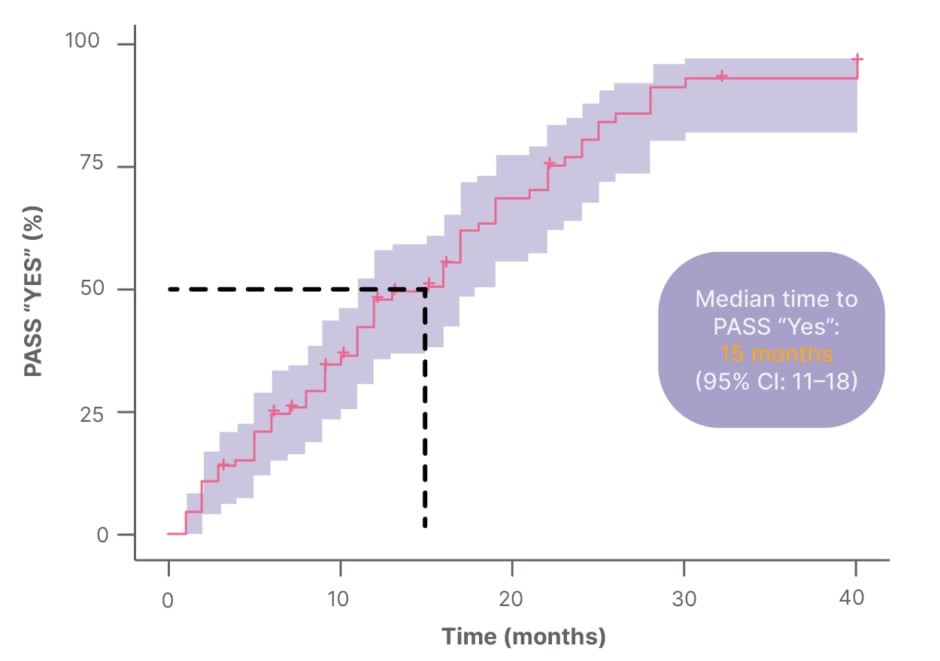
Figure 1: Time to Patient Acceptable Symptom State (PASS) “Yes” with Kaplan-Meier (n=67).11
In addition, the estimated thresholds for commonly used MG health scales, including the MG-ADL, the Quantitative MG Scale (QMGS), MG Composite (MGC), Myasthenia Quality of Life (MG-QoL15), and EuroQol 5-Dimension (EQ5D), all reflected patient-acceptable states, as per the PASS status.13
Time for Change
Vissing explained that the gMG field is “turning towards the potential of patient-centred assessment.” The MG-ADL, for example, is now a common primary outcome of MG clinical trials,14 and a recent consensus paper recommended the use of PASS following MG-ADL.15 “PASS is a wonderful key to the toolbox, if the patient answers ‘no’, we open the toolbox and start investigating, whether they are depressed, whether they have symptoms, and so on. It’s a really good question, it takes no time at all, and it complements the MG-ADL,” Vissing said.
ABC of Current gMG Treatments
The fact that many patients still report poor QoL, despite the wide range of therapies available, shows there is a need for “better, quicker acting, more efficacious medicines”, Vissing said.
Outlining the current gMG treatment landscape, Wiendl explained that cholinesterase inhibitors, such as pyridostigmine, are usually prescribed as a first-line therapy and, in mild MG, can produce rapid relief of symptoms.16 However, most people living with the condition will also require immunosuppressive treatment to suppress autoantibody production.16 Wiendl explained that autoantibodies drive gMG pathophysiology by altering the neuromuscular junction, resulting in weakened signal transduction and impaired muscle contraction.17,18 Depending on the type of gMG, the autoantibodies target key molecules at the neuromuscular junction, i.e., AChR, LRP4, or MuSK,18 and pathological changes in the thymus are believed to play a pivotal role in the pathogenesis of AChR+ MG.16 It is worth noting, however, that the pathophysiology of the disease is not yet fully understood in seronegative patients.18 Wiendl said: “We do not know if that is a sensitivity of detection issue, or whether there are other targets we have not yet recognised.”
Immune Therapy: Current Challenges and Future Potential
Immune therapy suppresses the overactivation of cellular elements of the specific and non-specific immune system. Current standard of care includes non-selective immunosuppressants, designed to inhibit or alter the immune response in the peripheral immune system.19 New treatment strategies, however, aim to selectively restore the body’s tolerance towards autoantigens, keeping the healthy immune system intact.20 “The Holy Grail of the new therapies is to alter the pathological process, and while not harming the rest of the immune system. To put it another way, if you eliminate the immune system, you would ‘cure’ MG, but the person couldn’t live,” Wiendl explained.
A number of immunosuppressive agents are currently available for the treatment of gMG. These include corticosteroids, azathioprine, mycophenolate mofetil, cyclosporin, cyclophosphamide, tacrolimus, and rituximab.21 Yet, such agents have highly variable latency of action, with azathioprine, for example, taking between 6–18 months, and corticosteroids 2 weeks, to elicit a response.21 Furthermore, they can present substantial risks of severe adverse side effects, Wiendl said, adding: “MG is one of the diseases that is very treatable, in principle, with just corticosteroids. But they are nasty in their mid- and long-term side effects.” Potential corticosteroid-related adverse events include Cushing’s syndrome, hyperglycaemia, hypertension, stomach ulcers, and myopathy, as well as skin atrophy/thinning, cataracts and glaucoma, osteoporosis, and osteonecrosis.22 Steroids are not the only MG treatment that carries side effect risk. Azathioprine, for example, can cause hepatoxicity, leukopenia, and nausea, and mycophenolate mofetil may lead to nausea, vomiting, diarrhoea, leukopenia, and opportunistic infection. Cyclosporine A’s potential side effects include hypertension, paraesthesia, and nephrotoxicity.23 Rituximab, an anti-CD20 antibody, works by depleting B cells, an effect that persists even after treatment discontinuation, and elicits a lasting change to the immune repertoire.23,24
New, targeted agents, such as C5 complement inhibitors and FcRn blockers, are emerging for more selective treatment of gMG. “These two groups of molecular compounds are changing the picture of neurological immunotherapy dramatically,” explained Wiendl. Complement inhibitors, which have been developed in the AChR+ population only, prevent C5a-induced chemotaxis of the proinflammatory cells, thereby preventing the formation of C5b-9-induced membrane attack complexes.25 This may prevent complement-mediated membrane damage at the post-synaptic membrane of the neuromuscular junction.25 FcRn blockers selectively target FcRn IgG recycling, lowering circulating IgG, including the pathogenetic autoantibodies.26 This, Wiendl said, may improve gMG manifestations. To date, the European Medicines Agency (EMA) has approved efgartigimod and rozanolixizumab,27,28 while two more FcRn blockers, nipocalimab and batoclimab, are currently in late-stage development.2,29 “These two groups of compounds [C5 complement inhibitors and FcRn blockers] have considerable advantages over almost all the other immunotherapies we are currently using,” said Wiendl.
Evolving Treatment Pathways
Evolving from immunosuppressive to immune-targeted therapy will require collaboration with the medical community, and a focus on how to optimise the evolving gMG treatment landscape. Ending the talk, Wiendl highlighted a proposed treatment pathway, published in last year’s German Guideline for the Management of Myasthenic Syndromes (Table 1), to help guide individual treatment decisions.30
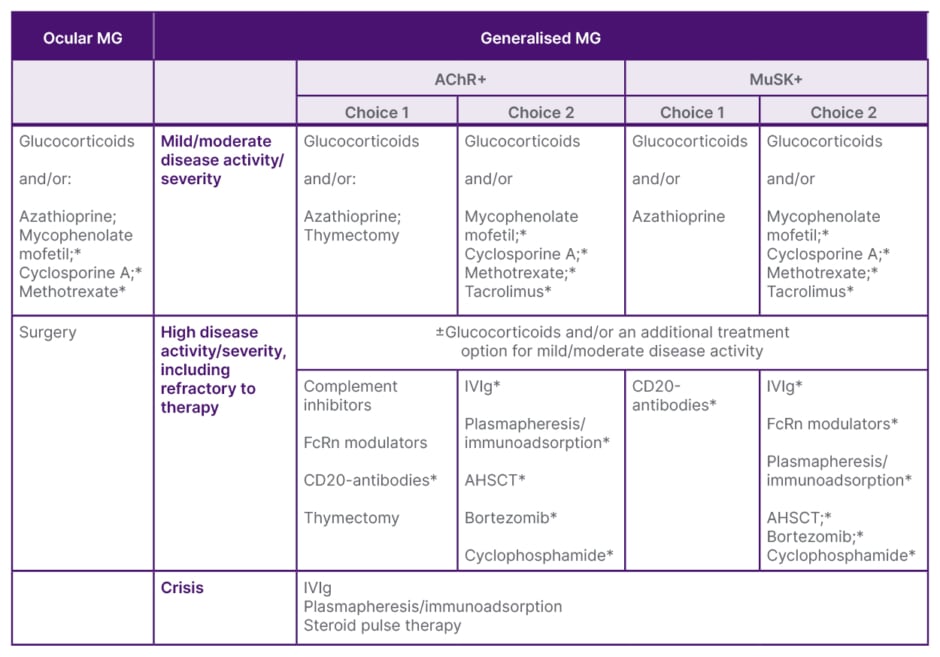
Table 1: Scheme for the disease-modifying therapy of myasthenia gravis.30
*Off-label therapies.
Ab: antibody; AChR+: acetylcholine receptor-positive; AHSCT: autologous haematopoietic stem cell transplantation; FcRn: neonatal Fc receptor; IVIg: intravenous immunoglobulin; MG: myasthenia gravis; MuSK+: muscle-specific kinase-positive.
Current and Future FcRn Blockers
In the last presentation, Claeys provided a more in-depth overview of FcRn blockers. FcRn, she explained, is a receptor that binds to IgG and protects it from degradation, prolonging its half-life.26 This can result in decreased circulating IgG levels, including the pathogenic autoantibodies that are responsible for gMG symptoms.26
Four FcRn have been developed for gMG. Efgartigimod, which is delivered via intravenous (IV) or subcutaneous (SC) infusion, is indicated as an add-on to standard therapy in adults with AChR+ gMG.27 Rozanolixizumab, which is delivered as a SC infusion, is indicated as an add-on to standard therapy for those with AChR+ or MuSK+ gMG.28 A Phase III trial of nipoclimab (NCT04951622), now completed, evaluated the agent, which is delivered via IV in AChR+, MuSK+, and LRP4+ patients with gMG. The trial was positive, meeting its primary endpoint (change of MG-ADL score from baseline to Weeks 22, 23, and 24).31,32 A Phase III trial of batoclimab (NCT05403541) is still ongoing in AChR+ patients, with a primary endpoint of change in MG-ADL score at Week 12.33 Each of these four FcRn inhibitors fall into one of two dosing pattern categories, namely cyclic, where patients undergo an initial cycle of treatment with any additional treatment cycle being based on clinical evaluation, or predictable (fixed dosing), where treatment cycles are administered weekly or every 2 weeks.
- Cyclic dosing:
- efgartigimod: once weekly for 4 weeks, with re-treatment being based on upon symptom resurgence and clinical evaluation27
- rozanolixizumab: once weekly for 6 weeks, with re-treatment being based on upon symptom resurgence and clinical evaluation28
- efgartigimod: once weekly for 4 weeks, with re-treatment being based on upon symptom resurgence and clinical evaluation27
- Predictable dosing:
- nipocalimab: once every 2 weeks32
- batoclimab: once every 1 or 2 weeks33
FcRn Inhibitors: The Latest Data
Claeys gave an overview of the currently available data on FcRn efficacy and safety, starting with the ADAPT Phase III study of efgartigimod, in which the primary endpoint was percentage of AChR+ patients who were MG-ADL responders in the first treatment cycle (8 weeks).34 A total of 84 patients received 10 mg/kg IV once weekly plus standard of care (SOC), while 83 received placebo plus SOC. Key inclusion criteria were gMG Myasthenia Gravis Foundation of America (MGFA) Class II–IV, with or without AChR antibodies; an MG-ADL score of ≥5; and receiving stable gMG treatment. Key exclusion criteria were rituximab or eculizumab in the previous 6 months, IV immunoglobulin (IVIg) or plasma exchange (PLEX) in the previous month, thymectomy in the previous 3 months, and pregnancy. At 8 weeks, 68% (44/65) of the AChR+ patients in the efgartigimod treatment group were MG-ADL responders, compared with 30% (19/64) in the placebo group (odds ratio: 4.95; 95% CI: 2.21–11.53; p<0.0001).34 The most common adverse events in the efgartigimod group were headache (29%), nasopharyngitis (12%), nausea (8%), diarrhoea (7%), upper respiratory tract infection (11%), and urinary tract infection (10%). These figures were broadly similar in the placebo plus SOC group. Rates of severe infection were low, at 1.2% (n=1) in the placebo plus SOC group, and 2.3% (n=2) in the treatment plus SOC group.34
The primary endpoint of the MycarinG Phase III study of rozanolixizumab was change in MG-ADL score from baseline to Day 43.35 Key inclusion criteria were MGFA Class II–IV, AChR+ or MuSK+, MG-ADL score ≥3, QMG score ≥11, and being considered for additional therapy, such as IVIg or PLEX. Key exclusion criteria were recent active/serious infection, severe oropharyngeal or respiratory weakness, and myasthenic crisis, defined as total IgG ≤5.5 g/L. Over 6 weeks, 66 patients were randomised to weekly rozanolixizumab at 7 mg/kg plus SOC, 67 to weekly rozanolixizumab at 10 mg/kg, and 67 to placebo plus SOC. Greater reductions from baseline in MG-ADL score were observed at Day 43 for both rozanolixizumab groups than in the placebo with SOC group, with least-squares mean differences from placebo of -2.59 (95% CI: -4.09 to -1.25; p<0.0001) for rozanolixizumab 7 mg/kg, and -2.62 (95% CI: -3.99 to -1.16; p<0.0001) for rozanolixizumab 10 mg/kg.35 There was one case (2%) of severe infection of COVID-19 pneumonia in the placebo plus SOC group, and no severe or serious infections in either of the rozanolixizumab groups. Headache was the most common adverse event, affecting 29 (45%) of patients in the rozanolixizumab 7 mg/kg plus SOC group, and 26 (38%) in the rozanolixizumab 10 mg/kg plus SOC group. In most cases, it was mild or moderate, with the more severe headaches being mainly recorded in the higher dose group.35
In the Vivacity-MG Phase III trial, 98 patients received nipocalimab plus SOC, and 98 received placebo plus SOC for 24 weeks.32 Key inclusion criteria were MGFA IIa-IVb, AChR+, MuSK+, LRP4+, or seronegative, and an MG-ADL score ≥6. Confirmed or suspected clinical immunodeficiency syndrome not related to gMG treatment and family history of congenital or hereditary immunodeficiency were the key exclusion criteria.31 Greater mean change in MG-ADL was seen with nipocalimab in seropositive patients with gMG over Weeks 22–24 (-4.7 points [standard error: 0.329]), with the difference of least-squares means compared with placebo plus SOC being -1.45 points (standard error: 0.470; p=0.002).32 The most common adverse events reported by the nipocalimab group were headache (14.3%), muscle spasms (12.2%), and peripheral oedema (11.2%), said Claeys. The rates of severe infection in this 24-week trial was 3.1% for the nipocalimab-treated patients, compared to 4.1% for the placebo group.”32
“Even though the FcRn inhibitors are lowering the IgG levels in the blood, it is clear that the rates of severe infection are very low,” Claeys said.
FcRn Inhibitors in Practice
In routine practice, there are potential benefits and advantages associated with both the cyclic and the predictable dosing patterns, Claeys noted. Sharing her personal opinion, she said: “In cyclic dosing, the benefit could be that the treatment cycles are administered based on the patient’s needs. For the predictable or fixed dosing, the benefits could be that this may provide a more sustained disease control. Potential drawbacks in cyclic dosing could be that you have to wait for a clinical deterioration before you can start a new cycle of treatment, and the treatment-free periods could be short. The potential drawback for predictable dosing could be logistical challenges, such as patients having to come into the clinic every 2 weeks.” More real-world evidence and clinical experience, she added, is needed to help physicians understand which FcRn blockers will be most useful in which patient groups going forward.
Summing up, Claeys said: “FcRn blockers are targeted, biological drugs that could be effective, well tolerated, and used in a broad population of patients with gMG. The different dosing strategies offer benefits and drawbacks for different patients, and this may allow for a more personalised approach to treatment.”






