
Meeting Summary
This symposium took place on Monday 24th June 2019, as part of the 2019 Peripheral Nerve Society (PNS) Annual Meeting in Genoa, Italy. Immune-mediated neuropathies such as Guillain–Barré Syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP), and multifocal motor neuropathy (MMN) are diverse inflammatory peripheral nerve disorders. International consensus guidelines recommend intravenous Ig (IVIG) as Level A for the treatment of GBS, CIDP, and MMN. Suggested induction doses of IVIG are 2 g/kg divided over 2–5 days, but maintenance doses are purposely less clearly defined and left up to the judgement of the clinician, depending upon the specific needs of the individual patient. Community-based neurologists treating patients with these rare inflammatory neuropathies may be unaware of optimal dosing regimens and patient response to treatment may therefore be inadequate. In this symposium, world-renowned experts in GBS, CIDP, and MMN shared their expertise and review of the literature to provide reasonable dosing regimens for neurologists who may rarely encounter these conditions.
Intravenous Immunoglobulin Dosing in Guillain–Barré Syndrome
Professor Bart Jacobs
The Dutch Guillain–Barré Study Group first published their experience with IVIG in GBS in 1992.1 In this study, the dosage employed, 0.4 g/kg/day for 5 consecutive days, was based upon previous experience in autoimmune diseases and it was uncertain whether it would be effective in acute GBS. The study showed that IVIG was at least as effective as plasma exchange in this clinical setting and was associated with less frequent complications. An important question arising from the study was whether 0.4 g/kg/day for 5 days is the optimal IVIG dose for all patients, or whether greater improvements could be achieved with a higher dose.
To investigate this question, a pharmacokinetic (PK) study was performed in which 174 patients with GBS received IVIG 0.4 g/kg/day for 5 days.2 After 2 weeks, there was a significant increase in serum IgG levels which then slowly declined (Figure 1A).2 Notably, there was a marked variation in PK profile. Patients were stratified into four groups according to the change in serum IgG levels and correlated with outcome; the proportion of patients who recovered the ability to walk unaided was largest in patients with the highest increase in IgG levels (Figure 1B).2
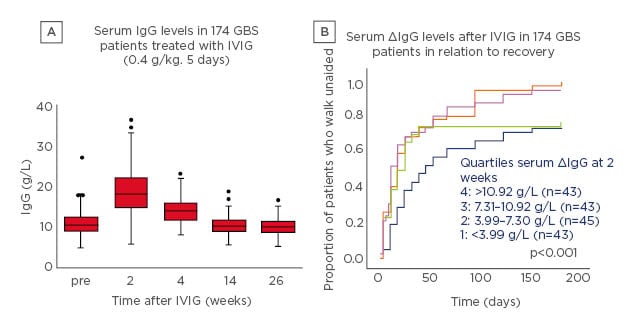
Figure 1: Intravenous Ig pharmacokinetics (A) and outcome (B) in patients with Guillain–Barré syndrome treated with intravenous Ig 0.4 g/kg/day for 5 days.2
5 Box: interquartile range; line in box: median; whiskers: range without outliers; dots: outliers.
GBS: Guillain–Barré syndrome; IVIG: intravenous Ig.
There are a number of questions concerning IVIG treatment of GBS regarding treatment failure, treatment-related fluctuations (TRF), treatment of mild GBS, treatment of Miller Fisher syndrome, and other variants.3
Treatment Failure
Treatment failure after IVIG 0.4 g/kg/day for 5 days is an important issue, especially if a patient has reached a stage requiring intensive care and respiratory support and shows no sign of recovery. When asked how to manage patients with apparent treatment failure after IVIG, neurologists at the PNS meeting most frequently opted to watch and wait. Other management choices, including an additional course of IVIG, methylprednisolone, or plasma-exchange, were frequently endorsed but there is a lack of evidence to support these choices. To help address this lack of evidence, a prospective observational cohort study (The International GBS Outcome Study [IGOS]) was designed to address clinical and biological determinants and predictors of GBS.4 The initial aim was to include at least 1,000 patients with a follow-up of 1–3 years using a web-based data entry system.5
In a recent update on the current practice of treatment, 1,023 patients were assessed including 743 with severe disease (unable to walk unaided), 168 with mild disease (able to walk independently), 70 with Miller Fisher syndrome, and 40 with other variants. Within these groups, 724, 126, 53, and 33 patients, respectively, were treated with IVIG, plasma exchange, or other immunotherapy. In the severe GBS group, 32% had no improvement with initial treatment and, of these, 35% received a second course of immunotherapy (Figure 2).5 In a nonrandomised, observational study in the IGOS cohort, the clinical course was compared in patients with a second course of IVIG and a predicted poor outcome. It was reported that 38/237 patients treated with a second IVIG course versus 199/237 given only 1 course showed no between-group differences in ability to walk unaided at 4 weeks.5 However, the groups were unbalanced because patients receiving a second course of immunotherapy were more severely affected.
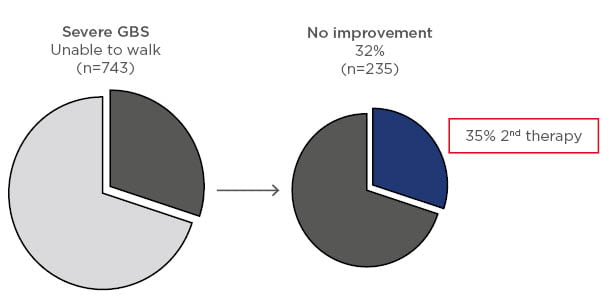
Figure 2: Response to first course of immunotherapy in patients with severe Guillain–Barré syndrome and percent of these patients receiving a second course of treatment.5
GBS: Guillain–Barré syndrome.
To resolve this issue, the Erasmus MC team in Rotterdam, the Netherlands, have initiated a placebo-controlled, randomised controlled trial (RCT) investigating the therapeutic effect of a second IVIG dose (SID-GBS trial),6 and the results are awaited with interest.
Treatment-Related Fluctuations
TRF are characterised by secondary deterioration after initial clinical improvement or stabilisation, and occur in up to 10% of patients with GBS.7 It has been hypothesised that relapses after treatment may occur when there is ongoing immune (re)activation (inflammation), resulting in a more protracted clinical course that is longer than the effective duration of action of immunotherapy.8 If the patient experiences >2 TRF, or if a TRF occurs >8 weeks after the onset of the illness, then a diagnosis of acute-onset, chronic inflammatory polyneuropathy, a variant of CIDP, needs to be considered.9,10 It has been reported that 3% of patients presenting with GBS have acute-onset CIDP.9 In these patients, maintenance treatment therapeutic options include IVIG or switching to an alternative therapy such as corticosteroids. It has been reported that relapsing patients respond well to a second course of immunotherapy.7 In the IGOS study, 53/1,023 GBS patients experienced TRF; of 50 patients initially treated with IVIG, 60% were retreated with IVIG, 8% switched to plasma exchange, and 32% received no treatment for their TRF. In patients who were re-treated for their TRF, the TRF occurred at an earlier time point than in untreated patients (median: 21 days versus 32 days; p=0.008).5
Treatment of Mild Guillain–Barré Syndrome and Variant Forms of the Disease
There does not appear to be any RCT with IVIG published on patients with mild or variant forms of GBS; however, based on the findings of the IGOS analysis it is clear that in current clinical practice the majority of patients with mild GBS, as well as those with variants such as Miller Fisher syndrome, receive immunotherapy (Figure 3).5
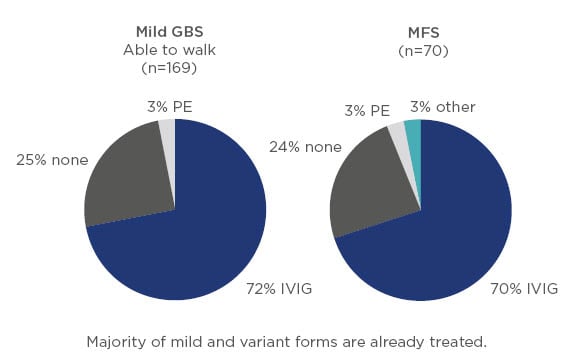
Figure 3: Treatment of mild Guillain–Barré syndrome and variant forms such as Miller Fisher syndrome.5
GBS: Guillain–Barré syndrome; IVIG: intravenous Ig; MFS: Miller Fisher syndrome; PE: plasma exchange.
Conclusion
Based on evidence to date, the optimal IVIG dose in patients with GBS is not well characterised, although guidelines recommend a dosage of 2 g/kg given over 5 days as the first-line treatment. This may be explained, at least in part, by a variability in IVIG PK properties in the patient population, resulting in inconsistent therapeutic responses. Consequently, neurologists are faced with several potential treatment dilemmas with GBS patients. This has led to variability in clinical practice and there is a need for additional clinical data from RCT to support the development of evidence-based treatment guidelines.
Intravenous Immunoglobulin Dosing in Chronic Inflammatory Demyelinating Polyneuropathy
Professor John England
CIDP is an acquired immune-mediated disorder of the peripheral nervous system. It is a heterogeneous, chronic, progressive disease that usually causes weakness, sensory loss, and neuropathic pain in the limbs.10 The efficacy of IVIG has been established in five randomised, placebo-controlled clinical trials, including the ICE study.11 This study, the largest reported trial of any CIDP treatment at the time, demonstrated the short-term and long-term efficacy and safety of IVIG and 10% caprylate-chromatography purified immune globulin intravenous (IGIV-C), and supports the use of IGIV-C as the standard for use in CIDP patients. This is despite there being very little evidence to support the relative benefits of different dosages and no true dose-response curves being established.
The PK parameters of IVIG are characterised by rapid attainment of peak levels, followed by a slow decrease in serum IgG levels with a mean/median half-life of 20–40 days as the Ig equilibrates between plasma and extravascular fluid. Equilibrium was achieved in 3–5 days.12 This extended half-life has been used to calculate a dose infusion frequency for IVIG of ‘every 3 weeks’. In terms of therapeutic effect, it is not known which IgG PK parameter helps define the best possible outcome (peak versus trough level, change in IgG level, steady-state level, etc.). The picture is further clouded by the fact that IgG can present in different forms (monomeric, dimeric, etc.) with various sialylation/glycosylation patterns that can impact terminal galactose and sialic acid residues.13 Better understanding of these factors may enable personalisation of dosing schedules.
European Federation of Neurological Societies (EFNS)/PNS guidelines recommendations on the management of CIDP with IVIG are based upon the findings of the ICE study: 2.0 g/kg as a baseline loading dose divided over 2–4 days, followed by maintenance infusions of 1.0 g/kg over 1–2 days every 3 weeks.14 However, this does not take into consideration patient weight status, or whether actual or adjusted bodyweight should be used. Consequently, IVIG dosing can vary widely and a recent USA survey reported that most practitioners used 0.4–1.2 g/kg every 2–6 weeks.15
Some key responses from qualitative interviews with external medical experts in the field of neurology in the USA regarding the treatment of CIDP are summarised as follows:
- IVIG was considered to be first-line treatment (approximately 70–90% of the time), because it was effective and had a better side-effect profile than steroids.
- However, if IVIG fails or is contraindicated, then steroids were the second-line choice.
- IVIG dosing: all experts gave a loading dose of 2 g/kg over 2–5 days which was typically followed by maintenance with 1 g/kg every 3–4 weeks. However, three respondents stated that for some patients this may be undertreatment and that they would freely give more IVIG, implying some disagreement about the correct maintenance dosage.
- No consensus on third-line therapy could be reached by the experts.
- All experts stated the need to establish expectations prior to treatment; any decisions to adjust dose will be based on strength and function only, and not on perceived sensory symptoms, such as tingling, pain, or fatigue. Patient agreement at the outset is considered important.
- All experts sought an objective response and relied heavily on clinical examination to determine treatment success or failure. In some cases, lack of improvement constituted failure, for others it was worsening of symptoms. However, one expert mentioned that if a patient had advanced CIDP, slowing disease progression may be an acceptable outcome.
- If there was a good response, experts tended to continue the maintenance dose until the patient achieved a maximal response and plateaued; then they started tapering the dosage. Experts typically felt that eventually they could wean up to 50% of patients. The preferred method for weaning patients off IVIG was by increasing the interval between doses.
- Two respondents incorporated a dose-reduction strategy to help maintain IgG levels at a steadier state (avoiding peaks and troughs).
- Diagnosing a patient as IVIG-dependent was rare, with most noting that they would resume a trial of weaning (i.e., cycle through the maintenance, weaning, and discontinuation process).
- When asked about the management of CIDP, expert neurologists indicated that community neurologists generally under or overtreated CIDP, with respect to dosing.
- Both over and underdiagnosis of CIDP are relatively frequent, and this can lead to problems with treatment.
- Undertreating CIDP is a concern because patients may be considered treatment failures, whereas giving a higher dose may have produced clinical improvement.
- Literature ‘wish list’ items included: dose weaning to a minimum tolerated dose, dose weaning to discontinue treatment (when/how to discontinue), and best treatment options for patients’ refractory to both IVIG and steroids.
- The idea of a validated patient outcome tool is appealing if it contains measures of strength, function, and disability.
- All experts mentioned using an ‘activities of daily living’ outcome tool, either Inflammatory Neuropathy Cause and Treatment (INCAT) Disability Score, Rasch-built Overall Disability Scale (RODS), Neuropsychological Impairment Scale (NIS), or other.
To evaluate how neurologists in community practice make decisions concerning the diagnosis and treatment of CIDP, Prof England’s group conducted an anonymous cross-sectional quantitative survey involving 100 practitioners in the USA.15 There was a wide variation in the use of specific guidelines to diagnose CIDP, with only 13% of respondents indicating that they used the globally accepted EFNS/PNS guidelines and almost 40% stating they did not routinely use a specific guideline. Many respondents had difficulty recognising the electrodiagnostic criteria used to diagnose CIDP or identifying atypical variants of the disease.
Regarding treatment, 44% of community practice neurologists chose IVIG and 20% indicated a preference for steroids; however, 24% of the respondents reported using steroids plus IVIG, despite guidelines stating that there is no evidence of clinical benefit for this combination. With respect to dosing, 67% of respondents indicated that they would use actual bodyweight, whereas 25% said they would use adjusted or ideal bodyweight, and 7% said they would start with a fixed dose regardless of the patient’s weight. For initial CIDP treatment, 55% of respondents used the recommended dose of 2 g/kg over 3–5 days. However, the remaining 45% of respondents quoted a wide range of doses from very low (0.01–0.04 g/kg) to unreasonably high (10.00–40.00 g/kg). Regarding maintenance treatment, the range was not as wide, but >40% neurologists used low (immune replacement) dosages of 0.01–0.50 g/kg. Most neurologists gave IVIG at least 3 months before deciding the response to treatment; however, 25% of survey respondents indicated that they treat their patients for 6 months. Finally, in terms of communication with the patient regarding weaning off IVIG therapy, responses were varied with no consensus. Most patients were not well-informed regarding this process and the timing of it. Others were simply told that they would be monitored and that treatment would be discontinued at some point in the future. Additionally, 21% of neurologists indicated that they made IVIG dosing decisions concerning the patient over the phone without a physical examination. To assess the patient’s overall condition, as well as strength, function, and disability, a consultation to ascertain how the IVIG is working is essential. There was no consensus on the duration of maintenance therapy prior to attempted weaning, and this is another area where more information is required to optimise overall patient management.
Conclusion
Based on the findings from a survey of 100 neurologists in community practice, diagnosis of CIDP remains problematic. Treatment of CIDP patients with IVIG is extremely variable, and under and overtreatment commonly occurs. Undoubtedly, CIDP is a difficult disease to manage because it is heterogeneous and there is a lack of good evidence in relation to various aspects of diagnosis and treatment. This should encourage the use of available evidence, perhaps in the form of an evidence-based clinical practice guideline that is brief, clear, and actionable. Ideally, IVIG dosage regimens should be derived empirically from high-quality studies that employed objective measures to ascertain the overall response to therapy.
Intravenous Immunoglobulin Dosing in Multifocal Motor Neuropathy
Professor Jean-Marc Léger
As way of background, a case of MMN involving a 46-year-old woman who presented with left foot drop was discussed. Over the next 5 years, she developed right hand weakness. At this stage, clinical examination revealed distal motor deficits of the left tibialis anterior and peroneus muscles, and in the distribution of the right median nerve. Tendon reflexes were normal except for the left ankle. There was no sensory or cranial nerve involvement and the Overall Neuropathy Limitations Scale (ONLS) score was 3. Electrophysiological studies confirmed conduction block in the right median nerve and denervation in the area of the fibula. Laboratory investigations showed that tests for antiganglioside antibodies (GM-1 and GM-2) were negative, the cerebrospinal fluid protein count (0.35 g/L) was normal, and MRI of roots and plexuses revealed 2 foci of hyperintensity on the right median nerve (proximal and distal), and hypertrophy and a hyperintensity in the left L2–S1 roots. At this stage, treatment was started with IVIG 100 g over 3 days, repeated every 4 weeks from June 2015 to January 2017. There was marked improvement of the motor deficits in the right hand, but lesser improvement in the left lower limb, and the patient relapsed 3 weeks after each infusion. In 2017, the patient relapsed 2 weeks post-infusion and electrophysiological studies identified a new conduction block in the left upper limb and ONLS was rated at 4.
Discussions with colleagues resulted in two suggested treatment approaches: the first was to maintain IVIG treatment, but to increase the dosage or reduce the duration between doses; the second was to add an immunomodulator to the current treatment regimen. Prof Léger and his team decided to continue with IVIG, but to reduce the dosing interval to every 3 weeks. The patient responded and has done well since this change in treatment with stable conduction blocks.
The challenges to Prof Léger’s understanding of MMN treatment are numerous and some questions that his group are wrestling with include:
Q1. Can we learn anything from the pathophysiology and natural history of MMN?
Q2. What are the best outcome measures?
Q3. What have we learned regarding short-term treatment with IVIG from prospective clinical trials?
Q4. What have we learned about long-term use of IVIG from retrospective and cohort studies?
Q5. How do we manage patients whose MMN is deteriorating despite maintenance therapy?
MMN is characterised by slowly progressive, asymmetrical, predominantly distal limb weakness, usually starting and predominating in the upper limbs, with no sensory loss and persistent conduction block. While the pathophysiology is still unknown, some elegant research is being performed in relation to peripheral nerve nodopathies that may be relevant.16 In recent years, there has been growing support for the concept that nerve dysfunction in MMN is probably related to autoantibody attack of proteins in the nodal, paranodal, and juxtaparanodal regions of the nerve, and this results in sodium ion channel dysfunction and conduction block. This may also explain why there is conduction block in MMN without true demyelination and a rapid initial response to IVIG.17
It is therefore important to identify the best outcome measures to use in MMN to guide repeat IVIG administration. Currently, there is no consensus on best outcome measures. A European Neuromuscular Centre (ENMC) workshop meeting proposed that the primary outcome measure in clinical trials should be the RODS for MMN, which has 25 selected items and is the only disease-specific outcome measure. This could be expanded in the future using the AMC Linear Disability Score (ALDS) or the ABILHAND questionnaire.1 However, at this stage it remains an exploratory outcome measure.
A Cochrane review evaluated 4 RCT19-22 that assessed the effects of IVIG on motor scales in 34 MMN patients.23 This analysis showed that strength improved in 78% of patients treated with IVIG versus 4% of placebo-treated patients. Disability improved in 39% of patients after IVIG treatment versus 11% after placebo (not statistically significant). Following the Cochrane review, a placebo-controlled RCT was published involving 44 adults with MMN randomised 1:1 to either double-blind treatment of IVIG followed by placebo for 12 weeks each, or the reverse. This study had the advantages of a longer follow-up duration (3 months) and disability assessment.24 Mean maximal grip strength of the more affected hand decreased 31.0% on placebo and increased 3.8% on IVIG (p=0.005). In 36% of participants, Guy’s Neurological Disability Scale (GNDS) scores for upper limbs worsened only during the placebo period, while 12% deteriorated only during IVIG treatment (p=0.021). A total of 69% of blinded patients switched prematurely from placebo to open-label IVIG, whereas <3% switched from IVIG to open-label IVIG (p<0.001). This RCT confirms the beneficial effects of IVIG in MMN, but further research is needed to investigate what level of improvement in disability can be achieved and its relative cost-effectiveness. EFNS/PNS recommendations for MMN treatment are that IVIG (2 g/kg given over 2–5 days) should be first-line treatment (Level A) when disability is sufficiently severe to warrant treatment, corticosteroids are not recommended, toxicity makes cyclophosphamide a less desirable option, if initial IVIG treatment is effective then repeat treatment should be considered in selected patients (Level C), the frequency of IVIG maintenance therapy should be guided by the response (good practice point), and typical treatment regimens for maintenance are 1 g/kg every 2–4 weeks or 2 g/kg every 1–2 months (good practice point).25 Globally, IVIG is considered a first-line treatment option for MMN.25-28
Since 2002, six observational studies have been published to provide information regarding the benefit of long-term IVIG treatment in MMN.29-34 The first of these studies reported that long-term maintenance (4–8 years) with IVIG had a beneficial effect on muscle strength and upper limb disability, but may not prevent a slight decrease in muscle strength. Electrophysiological results suggested a favourable influence of IVIG on remyelination and reinnervation in MMN patients, but axon loss could not be prevented.29 In Prof Léger’s study, 40 MMN patients were treated with IVIG and confirmed a significantly high short-term response.32 At the end of follow-up (mean 2.2 years), only 8 patients had significant remission and 25 patients (68%) were dependent on periodic (4–8 weekly) IVIG infusions. A similar conclusion was reached in a Dutch study involving 88 patients followed for a median of 6 years.33 Initially, 94% of patients responded to IVIG and nonresponders had longer disease duration before the first treatment. At the time of the analysis, 76% of patients were receiving maintenance treatment and the median dose increased from 12 to 17 g/week during follow-up. In contrast, a more recent Austrian study reported a reduction in IVIG dosage in MMN patients during long-term follow-up (mean: 7.5 years) from a mean of 1.8 g/kg at baseline to 1.1 g/kg at the last visit.34
Regarding the management of patients whose MMN is deteriorating despite maintenance therapy, EFNS guidelines recommend that if IVIG is not sufficiently effective then immunosuppressive therapy may be considered. However, no agent to date has been shown to be beneficial in a clinical trial and data from case series are conflicting. Preliminary data from uncontrolled studies show that a small number of patients improve on immunosuppressive therapy;35 however, RCT are needed to confirm overall efficacy and optimal treatment schedules. More recently, a Cochrane review investigated the use of immunosuppressants in MMN and concluded that mycophenolate did not significantly improve strength or function or reduce the need for IVIG. There were some reports of clinical benefit, but also serious adverse effects with cyclophosphamide, and there is very little evidence for drugs such as azathioprine, IFN beta, rituximab, or cyclosporine.36
Conclusion
Until definitive evidence is available for alternative treatments, IVIG remains the mainstay of treatment in MMN and there is a need to obtain the best outcomes with this agent.35 In the future, a better understanding of the pathogenesis and natural history of MMN may help optimise treatment approaches and possibly identify new therapeutic targets. Identification of the best outcome measures to predict clinical benefit with IVIG will also be important. Learning from the results of RCT, real-world retrospective studies, and extrapolation into clinical practice will be important for patients. There is a need to better understand factors associated with the stabilisation of MMN when patients are not being treated or their disease is worsening. Finally, the best candidates for long-term disease-modifying therapy need to be identified.






