
Abstract
Background: Tauopathies are a major group of neurodegenerative disorders characterised by the presence of tau-immunoreactive inclusions in the cytoplasm of neurons and glia. The spread of pathogenic tau along neuroanatomical pathways may play a significant role in the pathogenesis of neurodegenerative disorders. It is hypothesised that such a spread of tau along neuroanatomical pathways would give rise to a characteristic spatial pattern of the tau-immunoreactive neuronal cytoplasmic inclusions (NCI) in affected tissue.
Methods: The aim of this study was to investigate this hypothesis by comparing the spatial patterns of NCI in regions of the cerebral cortex in eight different tauopathies: Alzheimer’s disease, argyrophilic grain disease, chronic traumatic encephalopathy, corticobasal degeneration, frontotemporal dementia with parkinsonism linked to chromosome 17, Guam parkinsonism-dementia complex, Pick’s disease, and progressive supranuclear palsy.
Results: Regardless of disorder, tau isoform, or inclusion morphology, the NCI were most frequently aggregated into clusters, which were regularly distributed parallel to the pia mater. In many regions, the regularly distributed clusters of NCI range in size (400–800 µm) approximating to the dimension of cell columns associated with the cortico-cortical pathways.
Conclusion: The presence of regularly distributed clusters of NCI in the cortex of all eight tauopathies suggests an association between the pathology and the cortico-cortical pathways and is consistent with the pathogenic spread of tau along these connections. Hence, treatments designed to protect the cortex from this spread may be applicable across many tauopathies.
INTRODUCTION
The formation of inclusion bodies in the cytoplasm of neurons, also known as neuronal cytoplasmic inclusions (NCI), is a typical pathological feature of neurodegenerative disease.1-5 In many disorders, the inclusion bodies contain the microtubule-associated tau protein and are therefore referred to as tauopathies.2 Several disorders, both common and rare, are classified within this group, including the most common, Alzheimer’s disease (AD),6 as well as argyrophilic grain disease (AGD),7-10 chronic traumatic encephalopathy (CTE),11 corticobasal degeneration (CBD),12-14 frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17),15 Guam parkinsonism-dementia complex (GPDC),16 Pick’s disease (PiD),17 and progressive supranuclear palsy (PSP).18-20
The molecular composition of pathogenic tau varies between the different tauopathies. Tau is encoded by the tau gene on chromosome 17, and alternative splicing of exons 2, 3, and 10 results in six possible isoforms.21 Tau resulting from alternative splicing inclusion of exon 10 is known as 4-repeat (4R) tau, while tau lacking exon 10 is referred to as 3-repeat (3R) tau.2 Individual tauopathies are characterised by inclusions containing differing proportions of 3R and 4R tau. Hence, NCI in PiD are characterised almost exclusively by 3R tau;22 those in AGD, CBD, and PSP by 4R tau;1,4,23,24 and in AD, CTE, and GPDC, the inclusions have a more complex composition containing both 3R and 4R tau in differing proportions.16,25 FTDP-17 has a particularly complex tau pathology in which different individuals may predominantly exhibit either a 3R or 4R tau pathology and proportions of the two may vary. Different NCI morphologies are also present, the majority being either flame-shaped neurofibrillary tangles (NFT), predominantly found in AGD, AD, CBD, CTE, and FTDP-17, or globose, as present in the Pick bodies of PiD. By contrast, PSP may have both flame-shaped and globose NFT.26 Hence, NCI is a collective term for all NCI in the tauopathies, but there are also specific names applied to specific inclusions in certain disorders such as NFT and Pick bodies.
It is believed that there are relatively few cellular pathways contributing to cell death in neurodegenerative disease27,28 and, consequently, the tauopathies are likely to have pathological mechanisms in common. One common feature of these disorders may be the pathogenic spread of tau along neuro-anatomical pathways.29 Recent research suggests that several pathogenic proteins, including tau, α-synuclein, amyloid β (Aβ), and the disease form of prion protein, can be secreted from cells, enter other cells, and seed small intracellular aggregates within these cells.29,30 This raises the possibility, as first suggested by Hawkes et al.31 with specific reference to the synucleinopathy Parkinson’s disease,31 that pathogenic agents may be propagated through the brain along neuro-anatomical pathways. If pathogenic tau spreads from cell to cell in tauopathies, then the resulting NCI would exhibit a spatial arrangement pattern in the tissue that reflects this spread.32 Nonrandom distributions of the NCI have been observed previously in various tauopathies, which lends some support to this hypothesis.33-35 The objective of the present study was, therefore, to compare the spatial patterns of the respective tau-immunoreactive NCI in the cerebral cortex of eight different tauopathies (AGD, AD, CBD, CTE, FTDP-17, GPDC, PiD, and PS) and to answer two questions:
- Do the NCI exhibit similar patterns of spatial distribution across different tauopathies?
- Could these spatial patterns be the consequence of cell-to-cell spread of pathogenic tau along neuroanatomical pathways?
MATERIALS AND METHODS
Cases
Cases of AD (n=6; mean age: 78 years; standard deviation [SD]: 9.2), CBD (n=12; mean age: 90 years; SD: 9.7), PiD (n=10; mean age: 65 years; SD: 11.3), and PSP (n=8; mean age: 73 years; SD: 7.4) were obtained from the Brain Bank, Department of Neuropathology, Institute of Psychiatry, King’s College, London, UK.
AGD (n=25; mean age: 90 years; SD=9.7) and FTDP-17 (n=3; mean age: 77 years; SD: 4.7) cases were obtained from the Departments of Neurology and Pathology and Immunology, Washington University School of Medicine, St. Louis, Missouri, USA.
CTE (n=11; mean age: 70 years; SD: 6.4) and GPDC (n=3; mean age: 77 years; SD: 0.71) cases were obtained from Boston University Chronic Traumatic Encephalopathy Center, Boston, Massachusetts, USA (VA-BU-CLF Brain Bank). Cases were diagnosed using current consensus criteria for AD,36-39 AGD,40 CBD,41 CTE,11 FTDP-17,15 GPDC,42 PiD,15 and PSP.43
Tissue Preparation
All procedures performed in these studies were in accordance with the ethical standards of the Institute of Psychiatry, London, UK; Human Studies Committee of Washington University School of Medicine, St Louis, Missouri, USA; and the Institutional Review Board of Boston University, Boston, Massachusetts, USA, and were carried out according to the 1964 Declaration of Helsinki and later amendments. After death, the next-of-kin provided written consent for brain removal and retention for research studies. Brain tissue was preserved in buffered 10% formalin. Tissue blocks were taken from various cortical areas including the superior frontal gyrus (B8), the superior parietal lobule (B7), the inferior temporal gyrus (B22), the parahippocampal gyrus (B28), and the ambient gyrus (B27).
Brain material was fixed in 10% phosphate buffered formal-saline and embedded in paraffin wax. Immunohistochemistry was performed on 6–8 µm sections using various anti-tau antibodies (AT8, PHF-1, TP70). Sections were also counterstained with haematoxylin to reveal the various types of neuronal and glial cells and establish the tissue boundaries.
Morphometric Methods
In each region of the cerebral cortex the NCI were counted along a strip of tissue located parallel to the pia mater, using between 32 and 64, 50×250 µm sample fields arranged contiguously.44 The sample fields were located both in the upper (approximating to layers II/III) and lower (approximating to layers V/VI) cortex. The short edge of the sample field was orientated to be parallel with the pia mater and aligned with guidelines marked on the slide. Where cortical sections exhibited severe atrophy, as in PiD and CTE, only the upper cortical laminae were studied. The number of NCI present in each sample field was counted. NCI were measured in alkyltransferase rather than the argyrophilic grains for consistency with the other tauopathies, such as the NCI of AGD (tangles and pre-tangles), which are also characteristic features of the disease.7-10
Data Analysis
Changes in density of inclusions along the tissue parallel to the tissue boundary were analysed using spatial pattern analysis.45-47 This method uses the variance–mean ratio (V:M) to determine whether NCI were distributed randomly (V:M=1), regularly (V:M<1), or were clustered (V:M>1) along a strip of tissue. Counts of NCI in adjacent sample fields were then added together successively to provide data for increasing field sizes (e.g., 50×250 µm, 100×250 µm, and 200×250 µm) up to a size limited by the length of the strip sampled. V:M was then plotted against field size to determine whether the clusters of NCI were regularly or randomly distributed and to estimate the mean cluster size parallel to the tissue boundary. A V:M peak indicates the presence of regularly spaced clusters while an increase in V:M to an asymptotic level suggests the presence of randomly distributed clusters. The statistical significance of a peak was tested using t distribution.45 Spatial patterns of inclusions in the various regions were classified into four categories: random, uniform or regular, regularly distributed clusters, and large-scale clusters without evidence of regular spacing. The frequency of regions in which regularly distributed clusters were in the size range 400–800 µm (i.e., the size of the columns of cells associated with the cortico-cortical pathways) was also determined. A more complex spatial pattern was evident in some regions in which smaller clusters of inclusions were aggregated into larger clusters and the frequency of this spatial pattern was also determined. Comparison of frequencies among disorders was made using chi-square contingency tables tests. Mean cluster size of the NCI was compared among disorders using a one-way analysis of variance (ANOVA) (STATISTICA™, Statsoft Inc., Tulsa, Oklahoma, USA) followed by Tukey’s honestly significant difference post-hoc test. In each disorder, the correlation between cluster size of the inclusions, disease duration, and disease stage was tested.48-51
RESULTS
Examples of the spatial patterns exhibited by NCI in various tauopathies are shown in Figure 1. In AGD, the NCI (NFT) exhibited a V:M peak at a field size of 50 µm suggesting the presence of clusters of NFT, 50 µm in diameter, regularly distributed parallel to the pia mater. The V:M ratio of the NCI (NFT) in AD increased with field size, without reaching a peak, suggesting the presence of a large cluster of NFT at least 1,600 µm in diameter. The V:M ratio of the NCI (NFT) in CTE exhibited two V:M peaks at 50 µm and 400 µm suggesting clustering at two scales in the tissue.
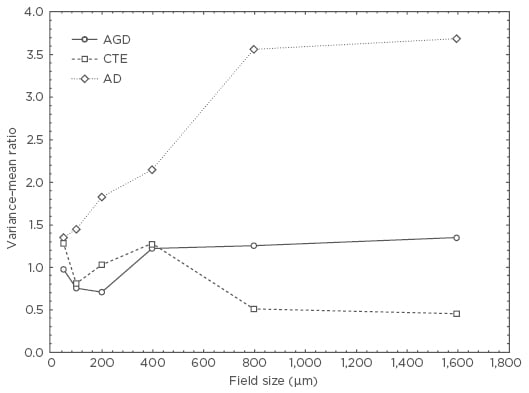
Figure 1: Pattern analysis plots showing examples of the spatial patterns exhibited by neuronal cytoplasmic inclusions in argyrophilic grain disease, chronic traumatic encephalopathy, and Alzheimer’s disease.
AD: Alzheimer’s disease; AGD: argyrophilic grain disease; CTE: chronic traumatic encephalopathy.
A summary of the frequencies of the four types of spatial patterns exhibited by NCI in the eight tauopathies is shown in Table 1. Most frequently, the NCI were clustered (mean cluster size in the range of 200–1,600 µm) and were regularly distributed parallel to the pia mater. This spatial pattern was present in all eight tauopathies but varied in frequency from 36% of CTE regions to 73% in AD. Larger-scale clustering of NCI, without evidence of regular spacing, was also present, especially in PiD and CBD. In some disorders, including AGD, CTE, and PSP, the NCI were also randomly distributed in a proportion of regions.
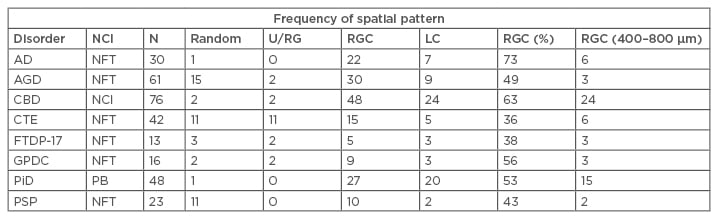
Table 1: Frequency of different types of spatial pattern exhibited by neuronal cytoplasmic inclusions in the upper and lower cortex in the various tauopathies.
Chi-square contingency tests comparing totals of upper and lower cortex. Comparing all taupathies: χ2=100.69 (21DF; p<0.001); comparing 3R tauopathies with 4R tauopathies: χ2=32.04 (6DF; p<0.001); 3R with 4R: χ2=12.81 (3DF; p<0.01); 3R with 3R and 4R: χ2=19.88 (3DF; p<0.001); 4R with 3R and 4R: χ2=12.54 (3DF; p<0.01); comparing FTDP-17 and CTE with idiopathic tauopathies: χ2=46.01 (6DF; p<0.01); FTDP-17 with CTE: χ2=1.40 (3DF; p>0.05); FTDP-17 with idiopathic: χ2=8.63 (3DF; p<0.05); CTE with idiopathic: χ2=45.31 (3DF; p<0.001); Comparing different NCI morphologies: All disorders: χ2= 36.96 (6DF; p<0.001); PiD with PSP: χ2=25.19 (2DF; p<0.001); PSP with all other disorders: χ2=17.33 (3DF; p<0.001); PiD with all other disorders: χ2=15.08 (3DF; p<0.01).
AD: Alzheimer’s disease; AGD: argyrophilic grain disease; CBD: corticobasal degeneration; CTE: chronic traumatic encephalopathy; DF: degrees of freedom; FTDP-17: frontotemporal dementia with parkinsonism linked to chromosome 17; GPDC: Guam parkinsonism-dementia complex; LC: large clusters; N: total number of cortical regions analysed for each disorder; NCI: neuronal cytoplasmic inclusion; NFT: neurofibrillary tangles; PB: Pick bodies; PiD: Pick’s disease; PSP: progressive supranuclear palsy; RGC: regularly distributed clusters; U/RG: uniform or regular.
Chi-square contingency table tests (Table 1) suggested that there were significant differences in the frequency of the various spatial patterns among tauopathies (χ2=100.69; 21 degrees of freedom [DF]; p<0.001), with AGD, CTE, and PSP having regions exhibiting a higher proportion of random distributions, and PiD and CBD a higher proportion of regions with large-scale-clustering.
The Chi-square contingency table tests also indicated that there were significant differences among disorders characterised by different tau isoforms (χ2=32.04; 3 DF; p<0.01) with 3R tau PiD having the lowest proportion of random or regular and uniform distributions compared with the 4R tau disorders (AGD, CBD, PSP) (χ2=12.81; 3 DF; p<0.01) and those characterised by both 3R and 4R tau (AD, CTE, and GPDC) (χ2=19.88; 3 DF; p<0.001). In addition, the 3R/4R combination disorders had a higher frequency of uniform distributions compared with both PiD (3R tau) (χ2=19.88; 3 DF; p<0.001) and 4R disorders (χ2=12.54; 3 DF; p<0.01).
There were also differences in the spatial patterns in disorders with predominantly flame-shaped NFT (AGD, AD, PSP, and CBD) compared with PiD, which has predominantly round or oval inclusions (χ2=25.19; 2 DF; p<0.001), and with PSP, which has both morphologies (χ2=17.33; 6 DF; p<0.001), with regularly distributed clustering being more frequent in PiD.
The Chi-square contingency table tests also suggested there were similarities in spatial patterns in cases characterised by a genetic aetiology (FTDP-17) or associated with head trauma (CTE) (χ2=1.40; 3 DF; p>0.05), but both differed from the remaining disorders which have a more uncertain aetiology in which regular-spaced clusters are less frequent. In a significant proportion of brain regions in all disorders, especially PiD and CBD, the regularly distributed clusters of inclusion were in the size range 400–800 µm (Table 1). A more complex spatial pattern, in which smaller clusters were aggregated into larger clusters, was also evident, with the exception of CTE and PSP.
Mean cluster size of NCI (Figure 2), averaged over cortical regions, varied among disorders (F=16.95; p<0.001). Post-hoc tests suggested that cluster sizes were greater in AD than in all the other tauopathies, cluster size was significantly larger in PiD than in the other disorders with the exception of AD, and that size was greater in CBD than in AGD, CTE, or GPDC. No statistically significant correlations were observed between cluster size of the NCI and either disease duration or disease stage in any disorder.
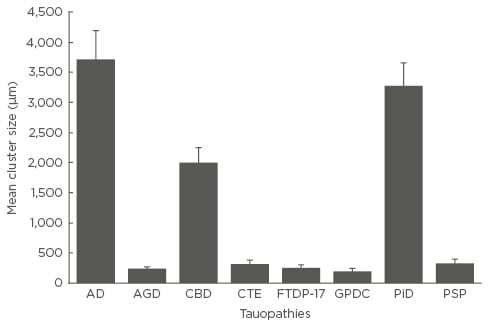
Figure 2: Mean cluster size of the neuronal cytoplasmic inclusions in the cerebral cortex of the eight tauopathies.
One-way analysis of variance (ANOVA) (with Tukey’s honestly significant difference post-hoc test) analysis showed a cluster size of F=16.95 (p<0.001). Significant differences shown between groups: AD larger than all other tauopathies; PiD larger than all others except AD; and CBD larger than AGD, CTE, FTDP-17, GPDC, and PSP.
AD: Alzheimer’s disease; AGD: argyrophilic grain disease; CBD: corticobasal degeneration; CTE: chronic traumatic encephalopathy; FTDP-17: frontotemporal dementia with parkinsonism linked to chromosome 17; GPDC: Guam parkinsonism-dementia complex; PiD: Pick’s disease; PSP: progressive supranuclear palsy.
DISCUSSION
There are a number of limitations to this study that should be considered when interpreting the results. The first point to consider is that different tau antibodies were used for the various tauopathies; however, no differences in density, type, or morphology of NCI have been observed between the tau antibodies used.33 A second consideration is that different anatomical pathways are affected in the different disorders and each of the cerebral cortex regions selected for study, mainly frontal and temporal lobe regions, may not reflect the critical regions in all disorders. Thirdly, different stages of disease progression are likely to be present within the various disorders. In an attempt to address the effect of disease stage, the correlation between cluster size of inclusions and disease duration and disease stage was investigated. The data, however, provided little evidence that cluster sizes altered consistently during the disease process, which could have been due to the small number of cases studied within each disease category. Finally, rare diseases, such as FTDP-17 and GPDC, were only represented by small numbers of cases and results for these tauopathies should be regarded as provisional.
The data suggest that NCI in the eight tauopathies, regardless of disorder, tau isoforms, aetiology, or inclusion morphology, were commonly clustered in regions of the cerebral cortex and, in a significant proportion of gyri, the clusters were regularly distributed parallel to the pia mater.34 However, the frequency of this spatial pattern varied among tauopathies, being most frequent in AD and least frequent in CTE and PSP. This spatial pattern was also present in disorders that had a prominent additional glial pathology (e.g., CBD35 and PSP26) or where an additional major molecular pathology was present, such as the Aβ deposits in AD. Variations in the overall frequency of different spatial patterns were also observed, the data suggest significant differences between AD, PiD, and CBD, in which regularly distributed clusters of inclusions were common and random distributions rare compared with AGD and PSP in which random distributions were significantly more common.
A number of features of the observed spatial patterns are consistent with the development of NCI in association with the neuro-anatomical pathways of the cerebral cortex and most specifically the cortico-cortical pathways.45,52 In cortical regions, the cells of cortico-cortical projection origin are clustered and occur in bands that are regularly distributed along the cortex. Individual bands of cells traverse the cortical laminae and, in primate brains, vary in width in the range of 400–500 µm up to 800–1,000 µm, depending on cortical region.53,54 Although there was considerable variation in cluster sizes among disorders, the width of the NCI clusters and their distribution along the cortex is consistent with an association with these pathways, with three exceptions. The first exception was that in some regions the NCI occurred in clusters larger than 400–1,000 µm, with some clusters being >3,200 µm in diameter, especially in AD, PiD, and CBD. In a small number of regions, clustering occurred at more than one site with smaller clusters of NCI aggregated together. The larger and more complex clusters suggest that the smaller regularly distributed clusters of inclusions could coalesce to form larger aggregations as the disease progresses, a process which appears to be a feature of AD, PiD, and CBD resulting in especially large clusters in these disorders.45 Another exception was that NCI were randomly distributed in some gyri, especially in AGD, CTE, and PSP. Random distributions are often the result of low densities of inclusions,34,55 which are likely to occur in the cortex in PSP, a primarily subcortical disorder.26 In AGD, by contrast, NCI may not be the most abundant tau pathology present in these cases, which are also characterised by significant aggregations of neuropil threads and argyrophilic grains.50,51,56
The size and distribution of the clusters of NCI in all the tauopathies studied suggested a close relationship in the cortex between the developing pathology and neuroanatomical pathways. The most likely explanation for this association is the spread of pathogenic tau among cortical regions along the cortico-cortical connections.29 However, differences in cluster size of the NCI were observed in different tauopathies, which suggests variation in the degree to which the cortical modules which comprise these connections were affected. The largest clusters were observed in AD, PiD, and CBD, while the smallest were found in AGD, CTE, and PSP. Differences in cluster size could be attributable to:
- Difference in density of the NCI.
- Differences in the vulnerability of anatomical pathways to the spread of pathogenic tau, a more selective group of neurons being compromised in AGD, CTE, and PSP.
- Differences in the rate of spread of pathogenic tau along neuroanatomical connections.
Propagation of pathogenic tau is not the only explanation for the results as they could represent intrinsic spatio-temporal-specific neuronal vulnerability affecting clusters of neurons and that not all stages in the proposed transfer have yet been demonstrated experimentally through cellular uptake, templated seeding, secretion, and overall transfer via synaptic and non-synaptic pathways.57
CONCLUSION
In conclusion, neurodegenerative disorders characterised by tau-immunoreactive NCI exhibit similar spatial patterns in regions of the cerebral cortex, consistent with their association with the degeneration of specific anatomical pathways. This association could reflect cell-to-cell spread of pathogenic tau in the tauopathies consistent with a common pattern of cortical neurodegeneration across disorders. Hence, it would be useful to trace the spatial pattern of inclusions along specific anatomical pathways in various tauopathies to test this hypothesis more rigorously. In addition, the data imply that different tauopathies may be amenable to similar interventions (e.g., immunotherapy that targets extracellular pathogenic tau) that could lead to its removal, thus preventing or slowing cell-to-cell propagation.58






