
Abstract
Background: Anti-N-methyl-D-aspartate receptor antibody encephalitis is an immune-mediated disorder characterised by a complex neuropsychiatric syndrome that often can be initially misdiagnosed. A small subset of the population is refractory to both first- and second-line therapies. These reasons make delays to the correct therapy a major concern, as early treatment may lead to better outcomes in children. Nevertheless, there is still benefit in additional medication courses despite a prolonged refractory state. The authors provide an illustrative case report and review of literature.
Case Presentation: The authors describe a 5-year-old female with 5 days of change in mental status; choreoathetoid movements were found to have positive anti-GluN1 antibodies in their cerebral spinal fluid. They failed first-line intravenous steroids and intravenous Ig and second-line rituximab, but then were discharged to rehabilitation without improvement over 3 months. Despite the time frame, they had a complete response to 12 sessions of plasma exchange with concomitant pulse steroids and subsequent intravenous Ig.
Conclusion: The authors’ case report and review of literature supports practices that prompt additional therapy for incomplete or failure of response in anti-N-methyl-D-aspartate receptor encephalitis despite prolonged symptom duration. Extended plasma exchange therapy may be beneficial in some treatment refractory cases.
Key Points
1. The authors present an illustrative case study of a commonly misdiagnosed complex neuropsychiatric syndrome, anti-N-methyl-D-aspartate receptor antibody encephalitis, in a 5-year-old patient.2. The patient presented with altered mood, abnormal movements, difficulty with motor control, and unintelligible speech. They were initially misdiagnosed with several differential diagnoses, including stroke, meningitis, and Sydenham’s chorea.
3. Early treatment can lead to better outcomes in paediatric patients; however, additional courses of medicine may provide benefit, and ought to be strongly considered even in cases of prolonged refractory state.
INTRODUCTION
Dysfunction of N-methyl-D-aspartate receptors (NMDAR) have been proposed as the aetiology for multiple neuropsychiatric diseases. While overactivity of NMDA receptors causing excitotoxicity has been implicated in epilepsy, dementia, and stroke, low activity may produce symptoms resembling those of schizophrenia.1 Autoimmune disruption to the NMDA receptors in anti-NMDAR antibody encephalitis is caused by the presence of cerebrospinal fluid (CSF) antibodies against the GluN1 subunit of the NMDA-R, leading to rapid deterioration in a patient’s psychiatric and neurologic state. The authors describe a 5-year-old female with typical presentation and positive anti-GluN1 antibodies, failing first-line intravenous steroids and intravenous Ig (IVIg) and second-line rituximab, but then discharged to rehabilitation without improvement over 3 months. Despite the time frame, they had a complete response to 12 sessions of plasma exchange with concomitant pulse steroids and subsequent IVIg.
CASE REPORT
A 5-year-old developmentally normal female, with no significant past medical or contributory family history, who was born at 27 weeks gestation, initially presented to an outside tertiary academic centre in autumn 2020 with 5 days of change in mental status and abnormal movements. Their mood had significantly tempered, yet they had continued superimposed episodes of inappropriate laughter, followed by difficulty with motor control, restless legs during sleep, and trouble grasping a pencil. Head CT was unremarkable, and serum white blood cell was elevated at 15.2×109 cells/L.
Their exam on presentation demonstrated prolonged periods of somnolence with rapid slurred and unintelligible speech, requiring significant stimulation to rouse. Non-suppressible choreoathetoid movements, characterised by intermittent abduction at the right hip and writhing movements with inversion at the right ankle, were present in the right lower extremity. Their left toe was upgoing.
The initial differential diagnosis included Sydenham’s chorea, autoimmune encephalitis, diffuse cerebral structural lesions, stroke, toxin exposure, metabolic derangement, and meningitis. Work up was normal or negative for urine drug screen, metabolites, head CT, and contrast enhanced brain MRI. Electroencephalography studies did not show epileptiform discharges or delta brush activity. For mildly elevated anti-streptolysin O titres of 400 IU/mL (anti-deoxyribonuclease B and throat cultures were negative, with a normal echocardiogram), they received 10 days of penicillin V for possible Sydenham’s chorea. Despite treatment, the encephalopathy progressed. Further CSF studies demonstrated a white blood cell of 14×109 cells/L, 90% lymphocytes, and negative CSF BioFire PCR (BioFire Diagnostics, Salt Lake City, Utah, USA) and aerobic cultures with gram stain. Encephalopathy autoimmune evaluation using cell-based assay confirmed NMDA-R antibody positivity in both serum and CSF, with titres of 1:2 in the CSF and 1:120 in the serum. An abdomen/pelvis MRI revealed a left ovarian focus of fat suspicious for teratoma, a possible paraneoplastic source for anti-NMDA-R encephalitis, but pathology following oophorectomy revealed normal ovarian tissue.
For worsening choreiform movements, increased agitation, and psychosis, the patient received IVIg 2 g/kg over 5 days and methylprednisolone 30 mg/kg daily over 5 days. They further received lorazepam, valproic acid (30 mg/kg/day divided in two doses), and clonazepam (1.75 mg/day in divided doses) for agitation and seizure prophylaxis. Given their lack of response during the first month after presentation, the decision was made to start second-line therapy, so the patient received 4 doses of weekly rituximab (375 mg/m2/dose). They continued to deteriorate with two prolonged episodes of unresponsiveness. A repeat brain MRI was unremarkable and repeat video electroencephalography showed only intermittent slowing. For worsening agitation and new onset nocturnal visual hallucinations, they received quetiapine (10 mg/kg/day in divided doses), which subsequently improved their sleep. Their clinical presentation deteriorated into a nonverbal state, with lack of eye contact and urinary incontinence, and extreme lethargy. They received a repeat course of 5 days IVIg (2 g/kg divided over 5 days) with pulse methylprednisolone (30 mg/kg/day). Second-line therapy cyclophosphamide was avoided due to concerns for infertility given the solitary remaining ovary. The patient was subsequently transferred to inpatient rehabilitation 1.5 months after symptom onset.
A second opinion was requested at 3 months post-symptom onset during inpatient rehabilitation. Consequently, they received 12 sessions of plasma exchange (PLEX), given every other day, with concomitant 30 mg/kg daily methylprednisolone and subsequent IVIg (2 g/kg divided over 3 days) after observing improvement following the second PLEX session. Repeat abdomen, pelvis, and chest MRI showed no evidence of malignancy. A brain MRI now demonstrated mild diffuse cerebral volume loss.
Following the sixth session, the patient demonstrated increased awareness of their environment, began speaking, and followed simple requests. Sedatives were tapered. One month follow-up post-hospital discharge, the patient made a complete recovery (Figure 1).
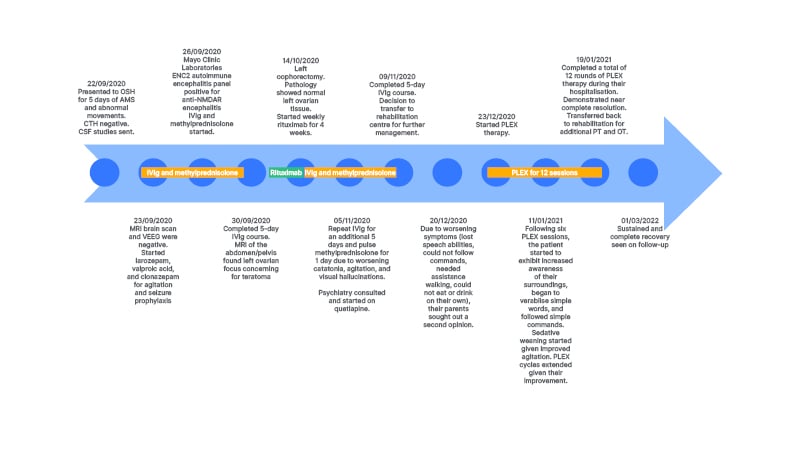
Figure 1: Patient treatment timeline.
AMS : altered mental state; CSF: cerebrospinal fluid; CTH: computed tomography of the head; ENC2: encephalopathy, autoimmune/paraneoplastic evaluation of spinal fluid; IVIg: intravenous immunoglobulin; LP: lumbar puncture; NMDAR: N-methyl-D-aspartate receptor; OSH: outside hospital; OT: occupational therapy; PLEX: plasma exchange; PT: physical therapy; VEEG: video electroencephalography.
DISCUSSION
Typical management of anti-NMDAR encephalitis involves the escalation of immunotherapy, starting with first-line therapies such as steroids, IVIg, and/or PLEX, and supplementing or transitioning to second-line therapies such as rituximab, cyclophosphamide, and, less commonly, azathioprine or mycophenolate mofetil as needed.2 In a large study by Titulaer et al.,2 almost half of the patients improved after first-line therapies. Of the patients who did not, nearly half improved after second-line therapies. Of the patients who remained refractory to both first- and second-line therapies, third-line therapy such as bortezomib, a proteasome inhibitor, or tocilizumab, an IL-6 receptor antagonist have been suggested.3,4 In other case reports, intrathecal administration of rituximab or methotrexate-dexamethasone with the rationale of improved CSF antibody removal has demonstrated success.5-7 Similarly, low dose IL-2, which aims to restore regulatory and effector T cells, has also been suggested for treatment-refractory anti-NMDA-R encephalitis.8 Smaller studies investigated daratumumab, an anti-cluster of differentiation 38 monoclonal antibody typically used in refractory myeloma treatment, and tofacitinib, a JAK inhibitor with good central nervous system penetration, and offered them as therapeutic options for refractory anti-NMDAR encephalitis.9,10
Numerous retrospective studies indicate a good prognosis, including fewer relapses, with early diagnosis and treatment of anti-NMDAR encephalitis.11,12 A large cohort study indicated that treatment delay greater than 4 weeks was associated with worse 1-year functional status.13 However, timely treatment is not always possible, given the complex presentation that may lead to psychiatric or non-autoimmune epileptic misdiagnoses, and lack of proven treatment options in refractory cases. Despite the common consensus that early diagnosis leads to better outcomes, some studies found no association between time to immunotherapy and outcome, suggesting that even delayed treatment could be of major benefit.14-17 One recent study indicated treatment delay did not affect neuropsychological outcomes, namely no long-term worsening of sustained attention, long-term verbal memory, or fatigue.18
Among second-line therapies, rituximab is believed still efficacious when initiated months or even years after disease onset. In a large multicentre retrospective study, 41% of 39 patients anti-NMDA-R failing first-line therapies had a definite benefit after receiving rituximab, on average, at around 1.2 months post-disease onset, but ranging from 0.05 to 5.10 years.19 Dou et al.20 studied eight children with refractory disease, where rituximab was given after a median disease duration of 57 days (range: 50.50–113.75 days). They found the use of rituximab led to a significant reduction in modified Rankin scale (mRS) and serum cluster of differentiation CD19+ B cells. Five patients (62.5%) had a good outcome (mRS≤2), including four patients (50%) who showed complete recovery (mRS=0) at last follow-up.20
The initiation of cyclophosphamide months from disease onset also has been shown to be beneficial in treating refractory disease. Kashyape et al.21 reported on two paediatric patients with refractory disease on steroids and IVIg, who had complete resolution of their movement disorders and a dramatic sustained clinical improvement of other symptoms in the domains of cognition, language, and behaviour following initiation of cyclophosphamide at Days 73 and 64 of symptom onset.
Similarly, among third-line therapies, treatment with bortezomib months to years after symptoms may also be beneficial in refractory disease. In a cohort study conducted by Scheibe et al.,22 five patients with refractory disease showed clinical improvement or disease remission despite delayed treatment with 1–6 cycles of bortezomib, initiated from 3 to 69 months since symptom onset. Another study reported that initiation of bortezomib 3 months into symptom onset in a 22-year-old female with refractory disease to both first- and second-line therapies showed rapid improvement in neurological deficits soon after the first injection.23 In a case report by Behrendt et al.,3 two patients who were severely affected with anti-NMDAR were treated with bortezomib at 7 months and 2.5 years from symptom onset, respectively, also showed significant clinical improvement.
Lengthier delays to efficacious therapy have been documented in several case reports. Sulentic et al.11 discussed that, despite delayed diagnosis and therefore immunotherapy ranging from 13 months to 8 years after initial presentation, three patients only had minimal residual cognitive deficits compared with their pre-morbid baselines. Another case report reviewed three patients with treatment refractory disease receiving tocilizumab 2–4 years post-symptom onset, with substantial recovery months after treatment initiation.4
The patient achieved complete remission on an extended course of PLEX therapy, an established empirical treatment with growing evidence for efficaciousness, at over 3 months post-symptom onset. PLEX likely filters harmful antibodies and proinflammatory cytokines to enable recovery in immune-mediated diseases.24 As a first-line therapy, PLEX is widely studied.24,25 In addition, in the paediatric population, there have been case reports suggesting remarkable success after PLEX therapy.26-28 A cohort study by Pham et al.24 identified that earlier initiation of PLEX therapy and PLEX followed by IVIg were able to provide the best outcomes for anti NMDAR encephalitis. In a large systematic literature review, Suppiej et al.25 identified the combination of PLEX, steroids, and IVIg to be the most efficacious option, with a mean of 7.3 exchanges in 62 patients.
The authors’ patient responded to a prolonged course of PLEX, eliminating the need for further costly inpatient rehabilitation. Although not well studied, longer courses of PLEX therapy in a number of small studies have emerged supporting its successful use. Simabukuro et al.28 described an 18-year-old with refractory anti-NMDAR encephalitis who failed first- and second-line therapies, but responded to 19 rounds of PLEX initiated on Day 45 after symptom onset.28 A report by Agrawal et al.26 described a 22-month-old infant who responded after 20 rounds of PLEX. In a review by Suppiej et al., the longest course of PLEX was 21 cycles.25 In another cohort study by Zhang et al.,25 the longest course of PLEX was 15 cycles in patients with refractory anti-NMDAR.29 The authors’ case report supports the mounting evidence that prolonged PLEX therapy should be a viable option when confronted with refractory disease.
Response to PLEX in the authors’ case was supported by the extended duration of time without improvement following initial hospital discharge. However, a potential delayed onset of action from rituximab could be postulated.
Nevertheless, this case study and review of literature highlight that delayed treatment must still be contemplated and offered to significantly symptomatic individuals prior to dismissal to in- or outpatient rehabilitation. This may be in opposition to other autoimmune disorders of the central nervous system, where classically the acute phase is considered concluded by 3 months. However, the distinction between subtle active disease and neuropsychological sequelae remains a challenging topic for this population.






