Abstract
Hereditary hemochromatosis is a condition resulting in tissue damage by pathological iron deposition due to genetic alterations. The clinical manifestations are diverse, and depend on the involvement of the affected organ. Complications such as cirrhosis, heart failure, diabetes, and arthritis are described. Iron deposition in brain tissues with neurological damage and presence of symptoms is not a usual finding. Some case series describe movement disorders as the clinical manifestation. The authors report a patient with movement disorder due to hepato-cerebral hemochromatosis, who showed clinical improvement after diagnosis and treatment. Hereditary hemochromatosis should be considered in the differential diagnosis of movement disorders in patients with signs of iron overload.
Key Points
1. Hereditary hemochromatosis is a condition that does not usually affect the nervous system; very few cases have been described that present with movement disorders.2. Unusually, hereditary hemochromatosis can present with a phenotype of cerebral iron overload, increased deposition in the basal nuclei, and associated parkinsonism.
3. Hereditary hemochromatosis should be considered in the differential diagnosis of movement disorders with pathological iron deposition in the brain.
INTRODUCTION
Hemochromatosis is a genetic disorder characterized by pathological tissue iron deposition with multiple organ involvement.¹ Hereditary hemochromatosis (HH) is one of the most common inherited disorders. The prevalence varies worldwide, and is more common in people from Northern Europe.² It is associated with tissue iron deposition, leading to functional alterations in several organs, with potential irreversible damage. This can lead to complications such as cirrhosis, heart failure, diabetes, arthritis, and a high risk of hepatocellular carcinoma.³ Nervous system involvement is uncommon, with a few reported cases manifesting as movement disorders.⁴ The authors present the case of a patient with movement disorders as the initial manifestation of hereditary hepato-cerebral hemochromatosis.
PATIENT INFORMATION
The patient was a 75-year-old male, with Type 2 diabetes and hypertension, without alcoholism. There was no family history of HH. He consulted for symptoms of 6 months of evolution characterized by tremor at rest, exacerbated after movements; loss of dexterity in both upper limbs; facial hypomimia; and rigidity and bradykinesia of the upper and lower limbs bilaterally, with increased support perimeter. The patient reported impairment in the ability to perform activities of daily living.
CLINICAL FINDINGS
Upon admission, the patient’s vitals revealed a temperature of 36.6 °C, a blood pressure of 130/75 mmHg, a heart rate of 76 beats per minute, a respiratory rate of 16 breaths per minute, and an oxygen saturation of 97% on room air. His cognition was preserved. Heart and lung auscultation had no alterations. The patient had a normal physical examination, except for rest tremor in both upper extremities. There was no ataxia. No significant alterations in pain sensitivity or sensitivity level were detected. In addition, the patient presented facial hypomimia, and rigidity and bradykinesia of the upper and lower limbs bilaterally, with increased support perimeter.
DIAGNOSTIC ASSESSMENT
Labs showed hemoglobin of 18.3 g/dL, hematocrit of 54.8%, leukocytes of 6,410 mm3, platelet count of 508,000 mm3, glycated hemoglobin of 6.3%, albuminuria/creatinuria ratio of 4.4 mg/g, and serum creatinine of 0.99 mg/dL. The brain CT scan was normal; therefore, a brain MRI scan was performed, which showed hypointensity in the echo gradient of the globus pallidus and midbrain, suggestive of iron deposition (Figure 1).
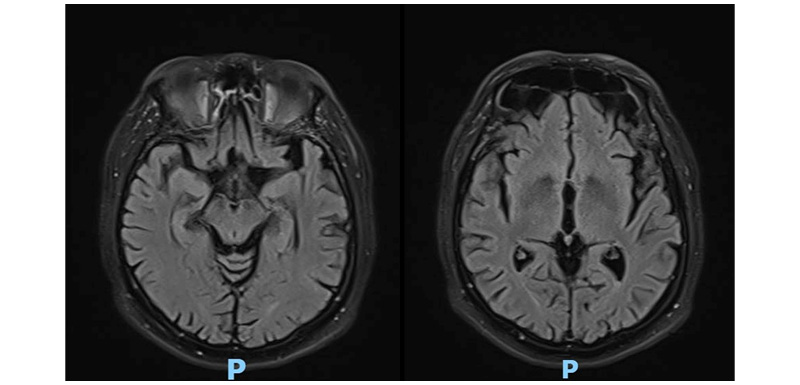
Figure 1: Brain MRI scan.
Complementary studies were performed, showing serum ceruloplasmin of 22 mg/dL, serum aspartate aminotransferase of 56 U/L, serum alanine aminotransferase of 26 U/L, total bilirubin of 0.2 mg/dL, normal thyroid-stimulating hormone, serum ferritin of 703.81 ng/mL, total serum iron of 255 ug/DL, transferrin saturation of 58%, hepatitis B surface antigen negative, antibody to hepatitis B core antigen negative, antibody to hepatitis C virus negative, and HIV negative. The study was complemented with hepatic MRI, which showed liver with decreased T2 signal and decreased signal in the phase sequence with R2* measurement of 95.4 s-1 ms, corresponding to intrahepatic iron concentration of 2.9 mg/g of dry liver by the Garbowski formula. Transthoracic Doppler echocardiogram was without segmental alterations, with good bilateral ventricular function, normal atrial dimensions, and normal valvular structures.
In view of the elevated ferritin and transferrin saturation levels, with the findings in the cerebral and hepatic MRI, a genetic panel was performed with positivity for homozygous HFE C282Y mutation, which confirmed the diagnosis of hereditary hemochromatosis.
THERAPEUTIC INTERVENTION
Management with phlebotomy was indicated, starting with induction of 500 mL in each extraction every 2 weeks, until serum ferritin lower than 50 ng/mL was reached. The phlebotomy protocol was continued every 4 months, and serum ferritin was monitored.
FOLLOW-UP AND OUTCOMES
After 3 months of treatment, improvement in neurological symptoms with functional independence was observed. Serum ferritin levels showed gradual decrease: 528 ng/mL, 374 ng/mL, 139 ng/mL, 45 ng/mL, and 34 ng/ml. The patient reported improved ability to perform activities of daily living.
DISCUSSION
HH is an inherited disorder characterized by increased dietary iron absorption with tissue accumulation, with potential for direct toxicity damage.⁵ The broad spectrum of tissue toxicity can result in diverse clinical manifestations, including cirrhosis, endocrine and exocrine pancreatic insufficiency, polyarthritis, skin hyperpigmentation, hypogonadism, adrenal insufficiency, hypothyroidism, and heart failure.²‚⁶
Iron is an essential element in various metabolic processes. In the brain, a sufficient amount of iron is particularly necessary for myelin synthesis, neurotransmitters, and energy production. Iron concentrations are highest in structures such as the globus pallidus, substantia nigra, putamen, and dentate nucleus.7 It has been suggested that excessive amounts of iron in the brain cause alterations in particularly susceptible tissues, leading to the generation of free radicals that exceed the capacity of cellular detoxification systems to scavenge them.8,9
The diagnosis of hemochromatosis is usually suspected in patients with elevated serum ferritin and transferrin saturation levels. Serum ferritin levels above 300 ng/mL in males, or 200 ng/mL in females, are usually considered elevated.5
Assessment of hyperferritinemia should include consideration of other possible etiologies, such as acute or chronic inflammatory states, liver disease, infectious diseases, and secondary iron overload. Certain environmental conditions, such as alcohol consumption, can also elevate ferritin.6 In the case presented, significant elevation of ferritin levels and elevation of transferrin saturation were found. On the other hand, there was no evidence of secondary causes of iron overload, nor a history of alcoholism that could predispose to the condition.
MRI findings suggestive of pathological iron deposition are characterized by a low T2-weighted signal. In fact, whenever available, iron excess should be confirmed by MRI with iron measurement protocol.10 In the present case, a decrease in the T2 signal and a drop in the signal in phase sequence with an R2* measurement of 95.4s-1 ms, corresponding to an intrahepatic iron concentration of 2.9 mg/g of dry liver by Garbowski’s formula, were observed.
Brain MRI typically shows T2 hypointensities and T1 hyperintensities, as well as low signal artefacts in susceptibility weighted imaging, mainly in the basal ganglia, dentate nucleus, and white matter.11 The case presented is notable for alterations in resonance imaging, especially in the globus pallidus and midbrain. MRI alterations may also be present in asymptomatic patients.12
Coexistence of hemochromatosis with diabetes and liver disease is rare, except in the presence of chronic alcoholism.3 Fracanzani et al.13 reviewed a prospective cohort of patients with hemochromatosis diagnosed between 1976–2007. Those patients with milder iron overload also had a lower prevalence of cirrhosis and extrahepatic manifestations, such as diabetes.13 The patient in question was diagnosed with diabetes 15 years earlier, with no history of alcoholism; it is uncertain whether iron deposition and tissue damage in the pancreas triggered the onset of diabetes in the present case.
In the general population, the allelic presentation of p.Cys282Tyr is estimated at 6.2%. The prevalence of the homozygous allele in patients with clinically recognized hemochromatosis is as high as 80.6%. Heterozygosity for the C82Y/His63Asp compound was reported in 5.3% of cases, and 19.4% of cases were unrelated to the gene.14
In the genetic panel performed in the clinical case presented, the presence of homozygous HFE C282Y mutation was confirmed.
Contrary to reports in the literature, the initial manifestation of hemochromatosis was neurological symptoms. The clinical case presented an iron overload phenotype with increased deposition in the basal nuclei and associated parkinsonism. Neurological involvement with movement disorders has been described in patients with HH, including parkinsonism, Parkinson’s syndromes, chorea, myoclonus, ataxia, dystonia, and tremor, with a mean age of onset at 57 years.4
The trend towards reversibility of neurological disorders with stabilization of iron stores through phlebotomy is highly suggestive of a causal relationship between HH and movement disorders. The patient showed significant improvement after 3 months of phlebotomy protocol.
CONCLUSION
This article presents a case of hereditary hemochromatosis due to homozygous mutation of HFE C282Y with movement disorder as initial manifestation. The presence of hepatic deposits was confirmed by MRI; however, a better clinical-radiological correlation was observed between the cerebral deposits and the patient’s symptoms. The favorable response to the phlebotomy protocol reinforces the premise of brain iron toxicity involvement. Hereditary hemochromatosis should be considered in the differential diagnosis of movement disorders in patients with signs of iron overload.
PATIENT PERSPECTIVE
Despite not being able to share a written perspective on the diagnosis, the patient expressed his bewilderment after the explanation of the atypical clinical presentation of his condition. The patient expressed full agreement with phlebotomy treatment, especially after noticing clinical improvement. On outpatient follow-up, he reported improved ability to perform activities of daily living. He continues to be followed up by the treating medical team.






