
Abstract
Behçet’s disease is an idiopathic chronic relapsing multisystem vascular-inflammatory disease of unknown origin, which usually presents with orogenital ulceration and uveitis and is identified as the triple-symptom complex. Primary neurological involvement in Behçet’s disease is known as neuro-Behçet’s syndrome (NBS). Clinical findings and neuroimaging demonstrate that there are two major forms of NBS: a central nervous system inflammatory parenchymal disease, and a less common nonparenchymal form that involves the large extraparenchymal vascular structures, mainly the venous dural sinuses. Cranial magnetic resonance imaging (MRI) typically reveals brainstem lesions with parenchymal involvement and an occluded dural sinus may be seen in the extraparenchymal type. Cerebrospinal fluid studies typically indicate inflammatory changes in the parenchyma and increased pressure with extraparenchymal involvement. Drugs used for the preventive treatment of NBS include azathioprine, cyclophosphamide, and anti-TNF agents.
INTRODUCTION
Behçet’s disease (BD), originally described in 1937 by the Turkish dermatologist Hulusi Behçet1 as a distinct disease with orogenital ulceration and uveitis known as the triple-symptom complex, is an idiopathic chronic relapsing multisystem vascular-inflammatory disease of unknown origin. The disease affects many organs and systems, causing mucocutaneous lesions, eye inflammation, musculoskeletal problems, and major vessel vasculopathy. There can also be cardiac, pulmonary, and gastrointestinal involvement alongside nervous system involvement.2
Due to the lack of specific laboratory, radiological, or histological findings for BD, accurate diagnosis of BD depends on clinical features. Various sets of diagnostic criteria have been published, with general agreement on the importance of mucocutaneous and ocular manifestations. The diagnostic criteria may not be met in many patients upon initial assessment because symptoms do not all appear at the same time. According to the International Study Group for Behçet’s Disease classification criteria,3 a diagnosis of BD requires recurrent oral aphthous ulcerations in combination with two of the following: genital ulcerations, skin lesions, eye lesions, or a positive pathergy test (Table 1). The International Study Group for Behçet’s Disease criteria for classification of BD has a reliable diagnostic value, reaching a sensitivity of 92% and a specificity of 97%. Minor diagnostic criteria include arthritis or arthralgia, deep vein thrombosis, subcutaneous thrombophlebitis, epididymitis, and family history, along with gastrointestinal, central nervous system (CNS), or vascular involvement.3

Table 1: Criteria for diagnosis of Behçet’s disease.*
*For a definite clinical diagnosis of Behçet’s disease, the patient must have recurrent oral ulceration plus at least two of the other findings in the absence of any other clinical explanations; †The pathergy phenomenon is a nonspecific skin hypersensitivity that is almost specific for Behçet’s disease. It is performed by inserting an 18-g needle into the dermis of the forearm. The reaction is considered positive if either a papule, a pustule, or an ulcer forms at the puncture site within 48 hours. Presence of erythema only is considered negative.
The epidemiology of BD varies geographically, with a higher prevalence along the ancient Silk Road countries, extending from the Mediterranean region to Japan. This is coupled by a similar variation in the human leukocyte antigen (HLA)-B51, which is strongly associated with the disease in high-prevalence areas.4 The HLA-B51 allele, which is accepted as a genetic risk factor strongly associated with BD, is seen in 50–80% of BD patients and has a prevalence of 20–25% in the general populations of Silk Road countries.5
The usual onset of BD is in the third or fourth decade of life; however, although rare, onset in children has also been reported.6 While BD shows equal frequency between each sex, males have a more severe disease course.7
The aetiology of BD is unknown but clinical and laboratory data suggest that there is dysfunction of both the innate and adaptive immune systems, resulting in an exaggerated response to viral or bacterial insults.8 Debate is ongoing as to whether this hyper-reactivity is an autoimmune phenomenon or, as suggested by more recent data, an autoinflammatory phenomenon.7 The core histopathologic phenomenon in some cases is vasculitis, and a low-grade, chronic, nonspecific inflammation in others.9
NEURO-BEHÇET’S SYNDROME
The primary neurological involvement in BD is named neuro-Behçet’s syndrome (NBS). Neurologic manifestations have been reported in 4–49% of cases; however, when large series are studied, their prevalence remains between 3% and 9% of all patients with BD.10
The age of onset of NBS, when excluding paediatric cases, is usually late within the third decade of life, with the mean duration between the onset of BD and NBS being approximately 5 years. NBS is almost three times more frequent in men than women, with up to 6% of patients presenting with neurological involvement without fulfilling the International Study Group for Behçet’s Disease classification criteria for BD.11
The suggested Cerrahpaşa School of Medicine diagnostic criteria for NBS in a patient that fulfils the International Diagnostic Criteria for BD is the occurrence of neurological symptoms not otherwise explained by any other known systemic or neurological disease or treatment, and in whom objective abnormalities consistent with NBS are detected either on neurological examination and/or with neuroimaging studies, including magnetic resonance imaging (MRI) and/or abnormal cerebrospinal fluid examination.12 In 2014, Kalra et al.13 suggested NBS diagnostic criteria were adapted and modified by the International Neuro Behçet’s Advisory Group and recommended for the diagnosis of NBS (Box 1); however, neither of these criteria have been validated. Although definite vasculitis is rarely observed, it has been considered that CNS involvement is caused by vaso-occlusive angiitis with venous predominance.9,14

Box 1: International consensus recommendation criteria for neuro-Behçet’s syndrome diagnosis.
BD: Behçet’s disease; CSF: cerebrospinal fluid; ISG: International Study Group; NBS: neuro-Behçet’s syndrome.
Adapted from Kalra et al.13
Patients with BD may present with different neurological problems, related either directly or indirectly to the disease. Primary neurological involvement or neurological involvements directly related to BD include headache (migraine-like, non-structural), cerebral venous sinus thrombosis (CVST) (extra-axial NBS), CNS involvement (intra-axial NBS), arterial NBS, neuro-psycho-Behçet’s syndrome, peripheral nervous system (PNS) involvement, and subclinical NBS. Secondary neurological involvement or neurological involvement indirectly related to BD encompass neurologic complications secondary to systemic involvement of BD (e.g., cerebral emboli from cardiac complications of BD, increased intracranial pressure secondary to superior vena cava syndrome) and neurologic complications related to BD treatments (e.g., CNS neurotoxicity with cyclosporine, peripheral neuropathy secondary to thalidomide or colchicine).15
Although NBS may occur with variable neurological problems, clinical findings and neuroimaging demonstrate that there are two major forms of NBS: a CNS inflammatory parenchymal disease and a less common nonparenchymal form that involves the large extraparenchymal vascular structures, mainly the venous dural sinuses. These two types of involvement very rarely occur in the same individual and it is assumed that they have a different pathogenic mechanisms.15
PARENCHYMAL NEURO-BEHÇET’S SYNDROME AND INTRA-AXIAL NEURO-BEHÇET’S SYNDROME
The majority of patients (70–80%) with neurologic involvement due to BD present with parenchymal CNS involvement. This form is termed parenchymal NBS (p-NBS) or intra-axial NBS and commonly affects the brainstem-diencephalicregion.16 p-NBS typically follows a subacute clinical course and the presentation can be headache, cranial neuropathies, dysarthria, ataxia, or hemiparesis.
Cognitive-behavioural changes, emotional lability, a self-limited or progressive myelopathy with urinary sphincter dysfunction, and, to a much lesser extent, other CNS manifestations, such as extrapyramidal signs and seizures, have been reported. There are also a small number of cases that have reported cerebellar degeneration, isolated optic neuritis, or recurrent peripheral facial paresis. Optic neuritis is extremely rare in BD and most visual symptoms are due to ocular involvement.17
Onset of p-NBS is usually associated with flaring of systemic features of BD. Some patients may only have a single attack, with or without residual neurologic deficits, but most will have recurrences with further sequelae, and some will have secondary progression. A small number of patients may have a primary, progressive course. p-NBS is usually associated with a poor prognosis; approximately 50% of p-NBS patients are severely disabled within 10 years of being diagnosed with the disease.12 Asymptomatic p-NBS corresponds to abnormal neurological signs in a BD patient without neurological symptoms. The detection of abnormalities on neurophysiological studies, as well as by neuroimaging in asymptomatic patients, further suggests that the subgroup of patients with subclinical CNS and PNS involvement may not be so uncommon. However, the clinical and prognostic value of detecting abnormalities in such diagnostic studies in this subgroup of patients is currently still not clear.18
MRI is the gold-standard neuroimaging tool for the diagnosis of NBS. Parenchymal lesions are generally located within the brainstem, occasionally extending to the diencephalon, and less often are within the periventricular and subcortical white matter. Acute or subacute lesions are hypointense to isointense on T1-weighted images, commonly heterogeneously enhanced with contrast-enhanced T1-weighted images, and hyperintense on T2-weighted and fluid-attenuated inversion recovery images (Figure 1).
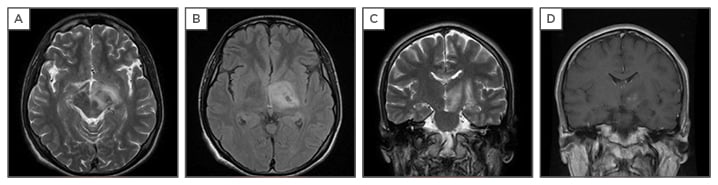
Figure 1: T1 and T2-weighted magnetic resonance imaging (MRI) of the brain.
A,B,C: Axial T2-FLAIR and coronal T2 images reveal hyperintense lesion mainly in the midbrain extending to diencephalon; D: Axial coronal T1 gadolinium sequence shows gadolinium enhancement at diencephalon.
Chronic lesions tend to be isointense and smaller, and it is not uncommon to see brainstem atrophy and third ventricular enlargement on follow-up MRI. In some patients with residual brainstem and subcortical lesions, images may rarely mimic multiple sclerosis. All these findings support the venous onset of the lesions.16
There are also a number of reports of NBS cases in which MRI images have shown mass lesions that mimicked brain tumours, some necessitating histological diagnosis.14 The presence of brainstem atrophy, particularly in the midbrain tegmentum and pons, have also been reported and correlated with a progressive form of the disease.19 Heterogenous enhancement with gadolinium may be seen in acute parenchymal lesions. The number of lesions detected with susceptibility-weighted imaging is larger than the conventional T2* gradient-echo.20 Most of the lesions in p-NBS were found to be haemorrhagic, supporting the proposed venous pathology.20
Spinal cord involvement is not common but does exist. In reported cases, the major site of involvement was the cervical spinal cord with the myelitis-like inflammatory lesions continuing for >2 segments and extending to the brainstem in some cases.21 The authors have observed a number of NBS patients presenting with longitudinal extensive myelitis in whom neuromyelitis optica-IgG was negative, and, recently, a distinct MRI pattern was identified which was labelled as Bagel Sign, supporting the venous pathology of BD.22,23
NON-PARENCHYMAL MANIFESTATIONS OF NEURO-BEHÇET’S SYNDROME
Cerebral Venous Sinus Thrombosis (Nonparenchymal Syndrome): Extra-Axial Neuro-Behçet’s Syndrome
The second most common form of neurologic involvement is cerebral venous sinus thrombosis (CVST), which may be seen in up to 12–20% of the patients with BD who have primary neurological involvement.12 This form is also termed vascular-NBS or extra-axial NBS. Clinical manifestations resulting from thrombosis of the intracranial venous system vary according to the site and rate of venous occlusion and its extent.
Our observation is suggestive that CVST in BD evolves gradually, so that a fulminating syndrome with violent headache, convulsions, paralysis, and coma is uncommon. Papilloedema and sixth nerve paresis are the most common clinical signs reported related to intracranial increased pressure.18
Cranial MRI usually shows an occluded dural sinus but otherwise normal parenchymal findings. In some cases, parenchymal lesions occur secondary to CSVT. MR venography might confirm the diagnosis and the extent of the CVST. According to the involvement, the superior sagittal sinus is the most common site of thrombosis, followed by transverse sinuses, deep cerebral veins, and cavernous sinuses, respectively.18 Venous haemorrhagic infarcts are not expected to occur constantly in people with CVST due to BD, in comparison to CVST caused by other aetiologies.
Arterial Neuro-Behçet’s Syndrome
Arterial involvement resulting in CNS vascular disease is rare, consistent with the systemic arterial involvement that is also uncommon in systemic BD. Arterial involvement affects mostly large arteries located at the extracerebral sites of the craniocervical arterial tree, suggesting that an extra-axial arterial pattern of NBS may exist, as well as an intra-axial arterial NBS pattern related to intracranial arteritis and intra-axial small arterial occlusions similar to the venous involvement seen in NBS. Aneurysm formation is common in the visceral arteries in BD but extremely rare in the intracranial or extracranial arteries.24
PSYCHIATRIC AND COGNITIVE DISORDERS
Anxiety and depression are the most common psychosomatic symptoms in BD. However, some patients with BD develop a neurobehavioural syndrome, which consists of euphoria, loss of insight or disinhibition, indifference to their disease, psychomotor agitation or retardation, paranoid attitudes, and obsessive concerns. The authors observed the development of these psychiatric symptoms either at the onset of other neurological symptoms of NBS or independently and unrelated to the use of glucocorticosteroids or any other therapy, and subsequently named this syndrome neuro-psycho-Behçet’s syndrome.25
HEADACHE
Headache is the most common neurologic symptom and occurs in 70% of BD patients. Headaches can result from different causes, including the nonstructural headache of BD, p-NBS, CVST, in association with ocular inflammation, and co-existing primary headaches (e.g., migraine or tension-type headache).
In a case series,26 it was observed that paroxysmal migraine-like pain occurred with exacerbation of BD systemic features. It may be explained by a vascular headache triggered by the immunomediated disease activity in susceptible individuals and may be seen in up to 18% of BD patients. This type of headache is not specific for migraine and similar headaches have been described in some other systemic inflammatory disorders, such as systemic lupus erythematosus.26
PERIPHERAL NERVOUS SYSTEM INVOLVEMENT
PNS involvement with clinical manifestations is extremely rare in BD. Mononeuritis multiplex, a peripheral neuropathy prominent in the lower extremities; polyradiculoneuritis, a sensorimotor axonal neuropathy; and an axonal sensory neuropathy with recurrent episodes of myositis have been reported. Although primary involvement of PNS is rare in BD, it should be kept in mind that polyneuropathy may occur secondary to thalidomide or colchicine treatment as a side effect.18
SECONDARY NEUROLOGIC INVOLVEMENT
Neurologic complications secondary to systemic involvement of BD, such as cerebral emboli from cardiac complications of BD or increased intracranial pressure secondary to superior vena cava syndrome, are indirect neurologic problems seen in BD. CNS neurotoxicity with cyclosporine and peripheral neuropathy secondary to thalidomide or colchicine use are neurologic complications related to BD treatments.11
DIAGNOSTIC STUDIES
Blood Tests
No laboratory tests provide a definite diagnosis of BD. Although erythrocyte sedimentation rate has been reported to be associated with BD activity, there is no defined relationship between elevated erythrocyte sedimentation rate or C-reactive protein and NBS activity. HLA testing can support the diagnosis in populations in which the disease is associated with the HLA-B51 phenotype and may help in the differential diagnosis. Despite being one of the diagnostic criteria, the pathergy test has a low sensitivity. According to the International Neuro Behçet’s Advisory Group’s consensus recommendations, a positive pathergy test in a patient with suspected BD and systemic BD features contributes significantly towards the diagnosis; however, a negative test does not exclude NBS.13
Cerebrospinal Fluid
Cerebrospinal fluid (CSF) pathology is found in 70–80% of patients with CNS involvement in NBS. If performed during the acute stage, CSF studies usually show inflammatory changes in most cases of NBS with parenchymal involvement. An increased number of cells, up to 100/mL or more, with modestly elevated protein levels are expected in most parenchymal cases. When the spinal tap is performed in the acute stage, the increased cells are likely to show a neutrophilic predominance, but this is not always the rule and a lymphocytic prominence may also be seen. In later stages, cell numbers decrease and lymphocytes are almost always the prominent cell type.10
Oligoclonal bands are usually absent, not exceeding 15–20%.27 Elevated concentrations of IL-6 in the CSF of patients with both acute and chronic progressive NBS in relation to disease activity have also been reported.28
DIFFERENTIAL DIAGNOSIS
In countries with a high incidence of BD, all chronic recurrent uveal inflammations, especially panuveal inflammations, must be differentiated from BD. Patients should be examined and followed-up for other manifestations of this symptom complex. In patients who present with symptoms of intracranial hypertension and in whom neuroimaging reveals thrombosis in one or more of the cerebral venous sinuses, BD needs to be included in the differential diagnosis. The differential diagnosis of p-NBS contains multiple sclerosis, stroke in young adults, CNS vasculitis, neurosarcoidosis, CNS tuberculosis, brainstem glioma, high-grade astrocytoma, and primary CNS lymphoma.29
MANAGEMENT OF NEURO-BEHÇET’S SYNDROME
Treatment of Parenchymal Neuro-Behçet’s Syndrome
There are no controlled trials for the management of vascular, gastrointestinal, and neurologic involvement of BD.30 Treatment strategies for NBS mostly depend on the clinical experience of the neurologists involved. Treatment options include high-dose intravenous methylprednisolone pulses for 5–10 days, followed by a taper of oral prednisolone (1 mg/kg for up to 4 weeks, or until improvement is observed), and should be followed with an oral tapering dose of glucocorticoids over 2–3 months in order to prevent early relapses.18
After remission is induced, long-term treatment with immunosuppressive agents should be considered in patients with parenchymal CNS involvement because this form in most patients can be followed by a relapse or secondary progressive course and may result in significant physical and cognitive deficits leading to neurologic disability.
Colchicine, azathioprine, cyclosporine, cyclophosphamide, methotrexate, chlorambucil, thalidomide, IFN-alpha, and anti-TNF agents are among the drugs used for the preventive treatment of the systemic features of BD and have also been trialled for CNS involvement.
In neurologic involvement, azathioprine has shown a tendency to improve the long-term outcome of neurologic involvement in a large uncontrolled series. Cyclosporine is an effective treatment in BD patients who have eye involvement; however, physicians should be mindful of the higher risk of developing CNS disease under cyclosporine treatment and it should be avoided in patients with established NBS.
It has been shown that in patients with NBS who had ongoing clinical relapses on single or multiple immunosuppressants, a switch to infliximab prevented further relapses and stabilised disability.31 Seventy-four BD patients without NBS were put on infliximab for either arterial or eye involvement because of failure of other immunosuppressants, none of whom had developed NBS at the time of last follow-up.31 The efficacy of TNF blockades for patients with severe NBS and resistance to standard immunosuppressive regimens was also shown in another recent case series.32
The authors’ current approach in the treatment of NBS depends on the severity of the initial neurologic event, as well as the systemic manifestations of BD. Treatment is decided in consultation with the patient’s treating rheumatologist. If the patient has poor prognostic factors and frequent systemic symptoms, it is usual to start infliximab in the first-line, otherwise azathioprine 2.5 mg/kg per day is started with tapering oral steroids.
Treatment of Cerebral Venous Sinus Thrombosis
Venous thrombosis of BD is usually treated with either high or medium-dose steroids because it is accepted that clot formation in veins is caused by a low-grade endothelial inflammation, rather than hypercoagulability; however, anticoagulation is the primary treatment in systemic venous thrombosis and CVST of any aetiology. In CVST this approach still remains controversial as BD patients with CVST are more likely to have systemic large vessel disease, including pulmonary and peripheral aneurysms that carry a high risk of bleeding. The use of anticoagulation should be considered only after such possibilities have been ruled out. Recurrences of CVST, although uncommon, are possible in BD and, as these patients are also at a higher risk of developing other types of vascular involvement, long-term azathioprine is also recommended in some of these patients with CVST.29
PROGNOSIS
Brainstem or spinal cord involvement, frequent relapses, early disease progression, and high CSF pleocytosis are poor prognostic features for p-NBS. Initiation with severe disability, primary or secondary progressive course, fever at onset, relapse during steroid tapering, meningeal signs, and bladder involvement are sometimes associated with poor outcome. Sex-associated systemic features age at onset do not change the prognosis of NBS.10,12 Although the neurologic outcome is better in patients with CVST, these patients may have significant mortality and morbidity due to more severe systemic vessel involvement.29
CONCLUSION
BD requires a multidisciplinary approach that involves rheumatology, dermatology, ophthalmology, and neurology departments. Neurologic involvement in BD is summarised in Table 2. Parenchymal NBS affects the telencephalic–diencephalic junction, brainstem, and spinal cord, and these patients present with a subacute onset of severe headache, dysarthria, ataxia, and hemiparesis. The prominent clinical feature of CVST in NBS is severe headache that usually develops over a few weeks. High-dose intravenous methylprednisolone pulses for 7–10 days, followed by gradual oral tapering over 3–6 months, is used for acute episodes. Although there is no randomised trial for the long-term treatment, azathioprine or infliximab initiation, depending on the relapse severity and accompanying BD symptoms, has been used.
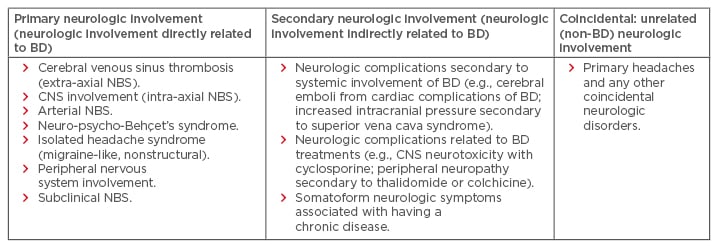
Table 2: The neurologic spectrum of Behçet’s disease.
BD: Behçet’s disease; CNS: central nervous system; NBS: neuro-Behçet’s syndrome.
Adapted from Siva and Saip.11





