Abstract
This review proposes an explanation for the pathogenesis of those neurodegenerative diseases which result in dementia and the resulting diversity of their disease phenotypes. The explanation is based on five principal observations, specifically: 1) neurodegenerative disease may be the direct consequence of neural ageing; 2) ageing may cause differential degeneration of neuroanatomical pathways; 3) breakdown of anatomical pathways may result in the formation of ‘reactive’ proteins; 4) these proteins may exhibit ‘prion-like’ behaviour and spread along anatomical pathways; and 5) neurodegenerative disease may be characterised by heterogeneity, overlapping phenotypes, and multiple pathology. The explanation proposes that genetic and environmental risk factors act cumulatively over a lifetime to increase an individual’s ‘allostatic load’, which determines the overall rate of neural ageing. This process results in the differential breakdown of neuro-anatomical pathways, influenced by their relative use or disuse during life, the consequence being the formation of one or more reactive proteins. Many of these proteins may spread through the brain from initial sites of ageing along neuro-anatomical pathways to affect specific neural networks. Variation in the proteins formed and in pathways of their spread result in the observed clinical and pathological diversity of disease phenotypes. Hence, minimising the factors that contribute to the allostatic load, together with cognitive and physical exercise to counter disuse of specific anatomical pathways over a lifetime, may be necessary to reduce the incidence of neurodegenerative disease.
INTRODUCTION
In 2015, 46.8 million individuals worldwide had a neurodegenerative disease, with 4.6 million new cases being recorded each year.1 Many of these neurodegenerative diseases result in dementia and it is these disorders that are largely addressed in this review. The overall prevalence of dementia, calculated by the European Dementia Meta-analysis (EURDEM) of all European studies, is 1.6% and 1.0% for males and females, respectively, in the 65–69 year age class, rising to 11.0% and 12.6% in the 85–89 year age class.2 Approximately 62% of dementia cases are attributable to Alzheimer’s disease (AD), 17% to vascular dementia (VaD) alone, 10% to a combination of VaD and AD, 4% to dementia with Lewy bodies (DLB), 2% for frontotemporal dementia (FTD), 2% for Parkinson’s disease dementia (PDD), and the remaining 3% of dementias for all other causes collectively.2,3
Normal physiological ageing often consists of the same changes in the nervous system that can be observed in neurodegenerative disease but at significantly reduced levels.4-7 Therefore, normal and pathological ageing results in brain atrophy and the formation of proteins in the form of ‘signature’ pathological lesions. Originally, the majority of neurodegenerative disorders were classified into two major molecular groups: 1) the tauopathies, including AD, Pick’s disease, argyrophilic grain disease, progressive supranuclear palsy (PSP), and corticobasal degeneration associated with the microtubule associated protein tau; and 2) the synucleinopathies, including PDD, DLB, and multiple system atrophy associated with the synaptic protein α-synuclein.8 Subsequently, cases that did not possess either tau or α-synuclein-immunoreactive inclusions were described. First, a proportion of FTLD cases were shown to have inclusions that were immunoreactive to the product of the transcriptor repressor gene (TARDP), specifically a transactive response DNA-binding protein of 43 kDa (TDP-43) (FTLD-TDP).9 Second, neuronal intermediate filament inclusion disease was shown to be associated with the product of the ‘fused in sarcoma’ (FUS) gene.10 Many of these diseases are therefore characterised by specific neuronal cytoplasmic inclusions, such as neurofibrillary tangles (NFT), and/or protein deposits such as the β-amyloid (Aβ) deposits in the form of senile plaques (SP) in AD and prion protein (PrPsc) deposits in Creutzfeldt–Jakob disease.
In most neurodegenerative disorders, there are small numbers of cases linked specifically to gene mutations and a larger number of sporadic cases not directly linked to genetics. Quantitative studies have demonstrated considerable similarities in the pathology of familial and sporadic forms of various diseases. Hence, variation in Aβ deposition was studied across several disorders including familial and sporadic AD using principal components analysis.11 Aβ deposition varied continuously across these disorders and did not distinguish between the familial and sporadic forms. In addition, there were no essential differences in the spatial patterns of Aβ deposits in familial and sporadic AD, both being distributed in regularly spaced clusters.12 There were no differences either in the spatial patterns of AD cases expressing or not expressing the apolipoprotein E (APOE) allele ε4, a major risk factor for AD.13,14 Furthermore, laminae distributions of Aβ deposits, which indicate the pattern of cortical degeneration, were similar in familial and sporadic AD, and the cortical layer at which Aβ deposits reached maximum density and the maximum density were also similar.
Similar results have been reported for familial and sporadic cases of FTLD with TDP-43-immunoreactive pathology (FTLD-TDP) although some differences in familial and sporadic FTLD-TDP have been reported. Hence, cases with and without progranulin (GRN) mutations have similar demographics, but GRN cases often have greater language deficits.15 Pathologically, cases lacking GRN mutations may have a less severe pathology affecting the neocortex and striatum.16 By contrast, a quantitative study of 94 cases of FTLD-TDP using principal components analysis suggested that the familial cases as a whole did not have a pathological phenotype that was distinct from the sporadic cases. In addition, the frequencies of the different laminar distributions in FTLD-TDP associated with GRN mutations17 were similar to those previously reported in sporadic FTLD-TDP,18 suggesting that the GRN mutations do not determine a specific pattern of laminar degeneration in FTLD-TDP. Hence, an explanation for neurodegenerative disease needs to explain the similarity of its familial and sporadic subtypes.
Given the present and future potential burden on health systems worldwide and the absence of widespread effective therapies, explanations are needed to account for the pathogenesis of neurodegenerative disease that collectively can lead to new treatment strategies. Based on interpretation of the literature, this review proposes an explanation of the pathogenesis of those neurodegenerative diseases resulting in dementia and attempts to account for the diversity of disease phenotypes. The explanation is based on five principal observations: 1) neurodegenerative disease may be the direct consequence of neural ageing; 2) ageing may cause differential degeneration of neuroanatomical pathways; 3) breakdown of anatomical pathways may result in the formation of ‘reactive’ proteins; 4) these proteins may exhibit ‘prion-like’ behaviour and spread along anatomical pathways; and 5) neurodegenerative disease may be characterised by heterogeneity, overlapping phenotypes, and multiple pathology.
THE FIVE OBSERVATIONS
Neurodegenerative Disease May be the Direct Consequence of Neural Ageing
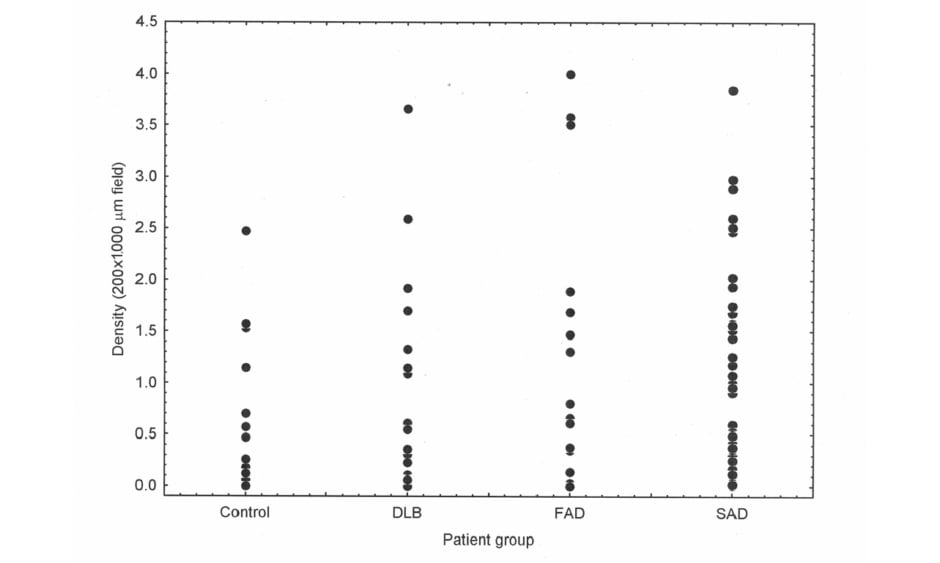
Epidemiological studies frequently agree that the greatest factor associated with neurodegenerative disease is age.4 In addition, in AD6 and PDD,5,7 there is direct evidence that neurodegeneration may be an accelerated form of ageing. Thus, most if not all AD neuropathological change (ADNC)19 also occurs in normal aged brains,20 including the enlargement of ventricles and loss of synapses and dendrites,21 together with the ‘signature’ histological features of AD (SP22 and NFT23,24). In addition, using Pittsburgh compound-B PET, a specific marker for Aβ deposition and therefore SP, Aβ was observed in 10–30% of healthy elderly patients.25 There is often considerable overlap in Aβ deposition in the normal elderly and in disorders such as AD and DLB (Figure 1), such that some control cases have greater densities of Aβ deposits than AD and DLB and some cases of dementia have very low densities. Other molecular markers of neurodegeneration also occur in a normal brain; phosphorylation and truncation of α-synuclein, which are characteristic of the ‘synucleinopathies’ Parkinsons’s disease (PD), PDD, DLB, and multiple system atrophy, are also normal events in the adult human brain.26 Moreover, in 110 cognitively normal individuals, 36% exhibited TDP-43,27 the pathological hallmark of a common subtype of FTLD.28 These data suggest that normal physiological ageing and neurodegenerative disease essentially share common cellular and molecular processes.
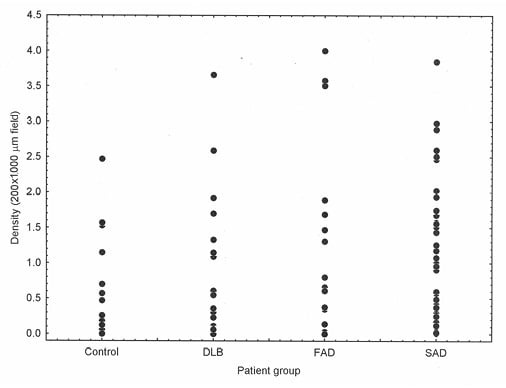
Figure 1: There is considerable overlap in the density of β-amyloid deposits between control and disease groups.
β-amyloid (Aβ) deposition in cases of normal elderly (control) brain, dementia with Lewy bodies, familial Alzheimer’s disease, and sporadic Alzheimer’s disease showing considerable overlap in the density of Aβ deposits between control and disease brains, between DLB and AD, and between FAD and SAD. DLB: dementia with Lewy bodies; FAD: familial Alzheimer’s disease; SAD: sporadic Alzheimer’s disease.
Ageing May Differentially Affect Anatomical Pathways
The efficiency of brain function depends on its long and short-range anatomical connections, there being fewer long-range connections as greater resources are required to maintain them.29 Normal adolescence is characterised by selective strengthening of the long-range connections, while ageing is associated with marked structural changes in the brain, including cortical thinning, degradation of myelin, and reduced connectivity. These changes especially affect the long-range connections, including those involving the basal forebrain, substantia nigra, locus caeruleus, and raphe nucleus.30 This reduced connectivity often results in a functional reorganisation later in life to compensate for the structural losses attributable to ageing.30 The pathways vulnerable to ageing include the structural covariance networks that subserve the language-related semantic network; the executive control network; the default-mode networks;31 the hippocampal network, which can affect memory function;32 and the resting state motor network.33 Hence, the selective disruption of anatomical pathways observed in different neurodegenerative disorders could result from the differential effects of ageing.34 Relative use or disuse during a lifetime could determine this selective disruption. Therefore, in individuals that suffer early blindness, there is significant reduction in white matter volume in the optic tracts, and radiation and significant loss of grey matter in the visual cortex.35 By contrast, physical activity may maintain or even restore pathways degraded by ageing.30,36-39
Breakdown of Anatomical Pathways Attributable to Ageing May Result in the Formation of ‘Reactive’ Proteins
Abnormally aggregated or misfolded proteins in the form of cellular inclusions have played a key role in the diagnosis, classification, and studies of pathogenesis.40 An important question is whether the deposition of abnormal proteins is a causal factor or a consequence of neurodegeneration.41 This question is controversial because protein aggregates may be either non-toxic (i.e., found in normal cells) or toxic, thus contributing directly to both primary and secondary phases of degeneration. The major evidence for a direct causative effect of aggregated proteins comes from studies of familial disease, their pathological phenotypes being similar, apart from age of onset, to those of sporadic forms of the same or related diseases.42,43 As a result, studies of gene mutation have had a major influence on the development of theories as to the pathogenesis of neurodegenerative disease as a whole. In familial disease, the major molecular constituent of a lesion is regarded as the residue of a direct or indirect effect of a pathogenic gene mutation that, via the accumulation of an insoluble protein aggregate, directly leads to cell death. This type of theory is best exemplified by the ‘amyloid cascade hypothesis’ proposed to explain the pathogenesis of AD, in which deposition of Aβ is the primary pathological event resulting in NFT, cell death, and eventually dementia.44
Nevertheless, a number of observations also suggest aggregated proteins are ‘reactive’ and a consequence of neurodegeneration. Firstly, the morphology and molecular constituents of cellular inclusions are dependent on cell type and location; cortical and subcortical NFT in AD, for example, comprise morphologically similar but antigenically different paired helical filaments.45 By contrast, cortical and brain stem Lewy bodies (LB) are morphologically different but antigenically similar,46 brainstem LB having an electron-dense core with radially oriented filaments differing significantly from cortical LB. Secondly, in cases of traumatic brain injury, amyloid precursor protein (APP) occurs in neuronal perikarya and in the dystrophic neurites surrounding Aβ deposits suggesting the production of APP as a response to neuronal injury.47 Specific neurons in the medial temporal lobe also secrete large quantities of APP and more APP-immunoreactive neurons present in these areas in cases of traumatic brain injury.48 Consequently, increased expression of APP after head trauma could be an acute-phase response to neuronal injury,49 with the overexpression of APP leading to increased deposition of Aβ. Thirdly, experimental damage to the nucleus basalis in rats decreased cortical choline acetyltransferase, elevated cortical peptides such as somatostatin and neuropeptide Y,50 and caused neuronal loss and the formation of SP in the cortex. Lesions of the nucleus basalis also elevated APP synthesis in the cerebral cortex suggesting a specific response to loss of functional innervation.51 Finally, the formation of NFT in AD may also be part of the neurons response to injury;52 thus, denervation of dopamine pathways and septal lesions affect both the cholinergic system and GABA neurons projecting to the dentate gyrus, and result in a loss of dendritic MAP2 and the appearance of tau-immunoreactive dentate gyrus granule cells.53
Proteins May Spread Along Anatomical Pathways
Studies suggest an association between neurodegenerative disease and the breakdown of specific neuroanatomical pathways.54 Populations of neurons lost in a particular disease often share a common metabolic abnormality and, therefore, neuronal connections between different regions could specify the pattern of cell losses in each disease.55 Research has confirmed these ideas and suggests that pathogenic proteins, including tau; α-synuclein, the disease form of PrPsc; and Aβ may be secreted from cells, enter other cells, and seed small intracellular aggregates within these cells.56,57 Pathological proteins, such as tau and α-synuclein, could exit cells via exocytosis or secretion and enter a new cell by endocytosis or by interactions with membrane lipids. Transfer may also occur via tunnelling nanotubes, which connect various neurons.57 Much of the support for pathological spread comes from in vitro experiments and there is less evidence from anatomical studies of neurodegenerative disease. However, if proteins spread from cell to cell in the cortex, the resulting inclusions may exhibit a spatial pattern that reflects this spread. Previous studies have suggested non-random distributions of the inclusions in the cerebral cortex in various disorders, the inclusions often exhibiting a distinct clustering pattern consistent with their spread via the cortico-cortical pathways.58
Neurodegenerative Disease may be Characterised by Heterogeneity, Overlap, and Multiple Pathology
Neurodegenerative disease comprises a wide diversity of clinical and pathological phenotypes.59 First, there is considerable variation in the severity and distribution of the pathology within many individual disorders, most notably in AD60 and FTLD.28,61 Second, many studies report ‘overlap’ between closely related disorders (i.e., coexistence of clinical and/or pathological features of more than one disorder in the same case).62 Third, many examples of more extensive ‘multiple pathology’ have been reported.63 In the parkinsonian syndromes, 38% of cases of PD have ADNC, 9% have PSP, 25% have argyrophilic grains, and 24% have congophilic amyloid angiopathy; in DLB, 89% have ADNC pathology, 1% have PSP, 21% have argyrophilic grains, and 25% have congophilic amyloid angiopathy.7 In addition, in a comparative survey of 1,032 cases representing ten different disorders, 361 cases (approximately 35% of the sample) were excluded because of multiple pathology.27 Multiple pathology is a consequence of either the co-occurrence of different pathologies by chance or the induction of one pathology by another. Hence, the coexistence of AD and PD is common because both disorders show a rapid increase in incidence with age and there is a high probability that both could coincide in the same individual.64 Alternatively, the presence of one type of pathology may encourage or induce the formation of another; e.g., the amyloid cascade hypothesis proposes that the formation of Aβ is the initial pathological event in the cascade directly leading to the formation of NFT.43
A POSSIBLE EXPLANATION
The explanation proposed in Figure 2 is that neural ageing is the initial trigger to neurodegenerative disease and is mediated by the ‘allostatic load’, i.e., the degree of lifetime stress experienced by an individual. The brain is the ultimate mediator of stress-related mortality through hormonal changes resulting in hypertension, glucose intolerance, cardiovascular disease, and immunological problems.65 Henderson4 also concluded that it was unlikely that genetic or environmental factors act directly, but that they accentuate some general process that occurs in the brain with age. It was postulated that the common feature associated with many of the risk factors is that they act by promoting the increasing liberation of oxygen free radicals, which exacerbates the rate of normal ageing, ultimately resulting in neurodegenerative disease. Second, this process results in synaptic disconnection and the differential breakdown of neuroanatomical pathways related, in part, to their degree of use or disuse during life. Third, synaptic disconnection results in the upregulation and deposition of various ‘reactive’ proteins such as Aβ, tau, α-synuclein, TDP-43, and FUS.51,66,67 Fourth, once a protein is formed, cell-to-cell transfer among interconnected neuroanatomical regions may occur, which results in recruitment of further pathogenic protein,56,57,68 as well as disruption of the blood–brain barrier resulting in an immunological response.
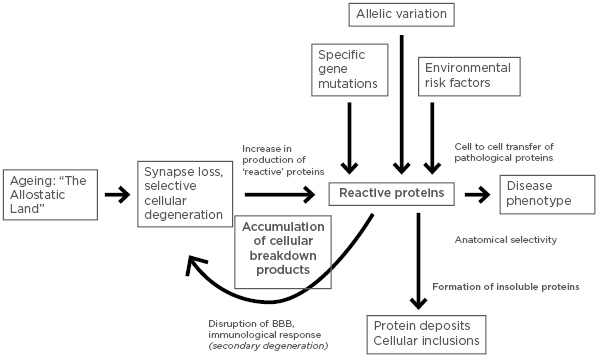
Figure 2: An explanation of the pathogenesis of neurodegenerative disease based on the five principal observations.
BBB: Blood–brain barrier.
The most overt manifestation of this process is in individuals with specific gene mutations that directly influence the outcome of age-related degeneration by determining the solubility and/or toxicity of the molecular products, and which rapidly overwhelm the cellular protection systems causing early-onset disease. By contrast, in individuals without a specific genetic mutation, the outcome is mainly soluble and smaller quantities of insoluble proteins that are degraded by the cellular protection systems and do not significantly accumulate to form pathogenic lesions until much later in life, causing late-onset sporadic forms of disease: often phenotypically similar to their familial counterparts.55 Variation in the observed disease phenotype results from differential vulnerability of specific neural pathways to the accumulating allostatic load and the effects of oxidative damage; genotypic variation, which determines the outcome of cellular degeneration and, therefore, the number, type, and frequency of proteins formed; and variations in the pathways of spread of various proteins along neuroanatomical pathways. The ultimate result of these processes is the complex overlap of many different pathologies, cases of neurodegenerative disease essentially forming a ‘spectrum’ or ‘continuum’ (Figure 3).59 In this hypothesised scheme, APOE genotype largely determines the degree of Aβ deposition with individuals expressing alleles ε2 or ε3 being associated with lower levels of Aβ deposition compared with those expressing allele ε4.69 Cases are further defined by which reactive proteins are formed: tau, α-synuclein, TDP-43, PrPsc, and FUS; each of these categories of disease may or may not be associated with Aβ depending on APOE genotype. Hence, cases in which Aβ and tau are predominant are ‘typical’ of AD and cases with tau deposition but little Aβ of the ‘classical’ tauopathies such as PSP, CBD, or AGD.
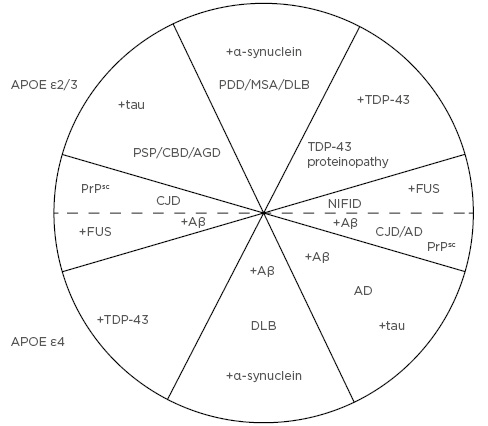
Figure 3: The ‘spectrum’ of neurodegenerative disease resulting from the proposed explanation in Figure 2.
Upper quadrants represent cases that express apolipoprotein E (APOE) genotypes ε2 or ε3 and are associated with low levels of β-amyloid (Aβ) deposition, while lower quadrants express APOE ε4 and are associated with significantly higher levels of Aβ deposition. Upper and lower quadrants are also defined by the formation of other major ‘reactive’ proteins, i.e., tau, α-syn (α-synuclein), prion protein, TDP-43, and FUS. Each of these categories of disease may or not be associated with Alzheimer’s disease neuropathologic change in the form of Aβ depending on APOE genotype. Hence, in cases located in the bottom right quadrant, Aβ and tau are predominant which is typical of Alzheimer’s disease, whereas cases located in the upper left quadrant are characterised by tau deposition but little Aβ typical of the ‘classical’ tauopathies, e.g., progressive supranuclear palsy, corticobasal degeneration, and argyrophilic grain disease. Boundaries between these groupings are unlikely to be distinct and there is continuous variation in disease phenotype both around the circumference and along the radii. AD: Alzheimer’s disease; AGD: argyrophilic grain disease; APOE: apolipoprotein E; CBD: corticobasal degeneration; CJD: Creutzfeldt–Jakob disease; DLB: dementia with Lewy bodies; FUS: ‘fused in sarcoma’; MSA: multiple system atrophy; NIFID: neuronal intermediate filament inclusion disease; PDD: Parkinson’s disease dementia; PrPsc: prion protein; PSP: progressive supranuclear palsy.
Predictions
First, this explanation predicts that many risk factors would be associated with neurodegenerative disease; in fact, any factor that can be shown to enhance the allostatic load is a potential risk factor. Several studies have confirmed this prediction, with the seminal review by Henderson,4 for example, identifying >20 different risk factors in AD, and several risk factors have also been identified in PDD.27 Second, there are individuals that reach considerable age without exhibiting neurodegenerative disease and, therefore, may represent a ‘survival elite’.4 The explanation predicts that such individuals would carry a low allostatic load. Third, as neural ageing is predicted to be the initial trigger of neurodegenerative disease, the effect of a gene mutation in transgenic experiments should be age-dependent, which has been demonstrated in a number of experiments.70-73 In addition, in the animal model for TgF344-AD, which incorporates mutant APP and PS1 genes, age-dependent amyloidosis, tauopathy, gliosis, apoptotic loss of neurons in the cortex and hippocampus, in addition to cognitive disturbance, was observed and may offer a more complete model of AD.74 Fourth, significant signs of neuronal degeneration as a result of ageing should precede the deposition of pathological proteins especially in sporadic disease. This statement is controversial and needs investigation because there are few current observations that indicate neuronal degeneration occurs prior to aggregated protein formation. Fifth, all ‘classical’ forms of neurodegenerative disease should exist with and without ADNC, a prediction already borne out by many disorders, such as AGD, CBD, Creutzfeldt–Jakob disease, DLB, PDD, and VaD, but less evident in PSP and MSA.62 Sixth, familial and sporadic forms of the same disease should have essentially the same phenotypes, an observation borne out by several studies.42,43
Limitations
The explanation presented has a number of limitations and also relies on controversial assumptions. First, there is limited data on the changes in brain connectivity with age leading to mild cognitive impairment and dementia. Second, there is limited evidence for the spread of aggregated proteins in cases of neurodegenerative disease, especially involving TDP-43 and FUS. Third, transgenic experiments do not always examine the influence of age on the developing pathology and, although pathological changes may be age-dependent, it is unclear whether they are a consequence of ageing as well as the genetic changes. Fourth, whether there are specific differences between familial and sporadic forms of the same disease is controversial. Fifth, whether the formation of aggregated proteins is a primary (‘causal’) or secondary (‘reactive’) process is an important element of the explanation and remains to be elucidated. The explanation assumes that the proteins are largely reactive and spread, ‘prion-like’, along neuroanatomical pathways. Further data on all these aspects are required to fully test the proposed explanation.
Implications
Given this explanation, it is less likely that many forms of neurodegenerative disease can be effectively treated by simple pharmacological intervention.75 Instead, the explanation suggests that attention should also be directed to reducing those factors that contribute to the life-time cumulative effects of allostatic load and oxidative damage52 and to encourage activity that contributes to exercising both cognitive and motor pathways throughout life. Reducing the allostatic load will require the identification of modifiable lifestyle and health-related variables to identify optimal combinations of such factors that could slow down the development of dementia.75 Current evidence is controversial and does not provide a sound basis for making specific recommendations and this question awaits further detailed study. The explanation suggests that exercise of a specific brain pathway may reduce the risk of a particular disease, such as cognitive exercise reducing AD and motor exercise PD. Some studies suggest that moderate intensities of physical activity over a lifetime may protect against volumetric brain loss most commonly affecting the prefrontal cortex and the hippocampus.37 In a further study, regular physical activity resulted in pathways less affected by typical age-related decline in cognitive function.19 In addition, individuals who exercised regularly reduced the risk of AD, the beneficial effect mediated by brain-derived neurotrophic factor acting on neuroplasticity and stress resistance,76 results not necessarily consistent with the explanation suggested in this review. In PD, however, there is evidence that heavy leisure-time physical activity may lower the risk of disease consistent with the hypothesis that continued exercise of the motor pathways may reduce their rate of aging.38 In addition, treadmill exercise in a murine model of PD improved motor performance and reduced α-synuclein expression while promoting tyrosine hydroxylase, dopamine transfer, and plasma dopamine levels.28 Thus, differential ageing resulting from variations in level of activity could be an important factor influencing the anatomical selectivity observed in neurodegenerative disease.39
CONCLUSIONS
Based on the literature, this review proposes an explanation of the pathogenesis of neurodegenerative disease and the diversity of its disease phenotypes based on 5 principal observations: 1) neurodegenerative disease may be the direct consequence of neural ageing; 2) ageing may cause differential degeneration of neuroanatomical pathways; 3) breakdown of anatomical pathways may result in the formation of ‘reactive’ proteins; 4) many of these proteins may exhibit ‘prion-like’ behaviour and spread along anatomical pathways; and 5) neurodegenerative disease may be characterised by heterogeneity, overlapping phenotypes, and multiple pathology. The explanation suggests that reducing the extent of the allostatic load over a lifetime and encouraging activity to exercise both motor and cognitive brain pathways may be necessary to reduce the incidence of neurodegenerative disease.




