Abstract
Diabetic kidney disease (DKD) has been an immense burden on the healthcare system, and is the leading cause of end stage kidney disease worldwide. DKD involves various intersecting pathways that lead to progressive kidney damage. Due to its versatile pathogenesis, DKD has been a formidable adversary. For many decades, there has not been much development in the arsenal in the fight against DKD, but recently, multiple new prospects have emerged due to the breakthrough in understanding of DKD pathology.
Tireless research of the changes occurring in the kidney as a result of diabetes, and the factors driving these changes, has led to the invention of medications that hopefully will be highly impactful in preventing end stage kidney disease in patients with diabetes. In this review, the authors summarise the timeline of the pathological changes that occur in DKD, the mechanism driving these pathological changes, and the recent discoveries in the pathways leading to DKD. These span over changes in metabolic pathways, inflammatory cascades, epigenetic alterations, and the description of their effects at cellular to structural levels in the kidney as a byproduct of uncontrolled hyperglycaemia. The authors also correlate these mechanisms with a few of the medications that are being utilised to slow down DKD, and some in the pipeline, with some references to the trials that support their use.
Key Points
1. Forty percent of patients with diabetes will have subsequent diabetic kidney disease (DKD). DKD involves various intersecting pathways that lead to progressive kidney damage, and is the leading cause of end-stage kidney disease worldwide.2. High glucose levels impact haemodynamics, hormone production, metabolic pathways, oxidative stress, and inflammation. DKD damages the podocytes, glomeruli, and the tubules, as well as causing changes on the cellular level including mitochondrial injury and epigenetic changes.
3. Improving understanding of pathological pathways of DKD has informed development of targeted therapies.
INTRODUCTION
It is imperative to understand the pathway in order to alter the course. Diabetic kidney disease (DKD) has been a challenge for nephrology for the last three centuries, despite the discovery of diabetes much earlier, around 1500 BC.1 A lot of progress has been made against the progression of DKD, but it still remains the most common cause of end stage kidney disease (ESKD) worldwide. In 2021, 9% of the healthcare budget in the USA was spent on diabetes, and the worldwide count rose to 537 million adult (20–79-year-old) patients.2 Forty percent of patients with diabetes end up with DKD and 10% of patients with diabetes end up on dialysis. Of the ESKD population, 60–80% are either diabetic, hypertensive, or both.2,3 In this review, the authors shed light on the recent advances in understanding the pathophysiology of DKD and summarise some of the correlating therapeutic interventions used across the globe.
TIMELINE OF DIABETIC KIDNEY DISEASE
Diabetes can go unnoticed and undiagnosed for years in the absence of appropriate screening. Similarly, it can take years before the effects of diabetes on the kidney can be picked up clinically, although the changes start soon after its onset. Even though DKD does not follow an exact timeline, patients with diabetes followed up for 10 years in the UK were noted to progress to microalbuminuria at a rate of 2% per year after the diagnosis of diabetes. Furthermore, the study noted that there was a 2.8% per year progression to macroalbuminuria from the microalbuminuria.4 Fourteen percent of patients developed renal impairment without preceding microalbuminuria.4 An autopsy study on 168 patients with diabetes noted that 63% of patients already had histological evidence of DKD, with almost 19% of those patients without any clinical evidence of DKD.5 Patients with Type 2 diabetes are less predictable, mostly due to inaccuracy of diagnosis, while patients with Type 1 diabetes have been noted to follow a more predictable pattern of kidney injury. During the first 5–10 years, epidermal growth factor receptor (eGFR) can overestimate the kidney function due to hyperfiltration followed by a decrease in eGFR and hypertension, usually noticed around 15 years after diagnosis,6 as described by the Mogensen classification of diabetic nephropathy. Increase in kidney size can start within 5 years of diagnosis with glomerular basement membrane expansion and mesangial thickening, taking about 10–15 years from onset. Overt fibrosis was noted after approximately 20 years of Type 1 diabetes.6 Despite this general pattern, the course of DKD remains irregular. In many other studies, it was also noted that the decline in eGFR in more than 50% of patients with diabetes did not follow a linear pattern.
PATHOPHYSIOLOGY
Diabetes, a name coined from passing a lot of urine and mellitus (meaning sweet) speaks itself of uncontrolled blood glucose levels. The end result of uncontrolled hyperglycaemia in the kidney is nephron death. This manifests in the form of glomerulosclerosis, interstitial fibrosis, and tubular atrophy, but there are a lot of milestones and instigators along the way. Fibrosis is the end point of years of micro- and macroscopic changes resulting from the diabetic milieu, as noted in Figure 1. The inception of the renal involvement in this disorder is the onset of uncontrolled levels of blood glucose. The authors will discuss the framework that stems from hyperglycaemia, and concludes with renal fibrosis.
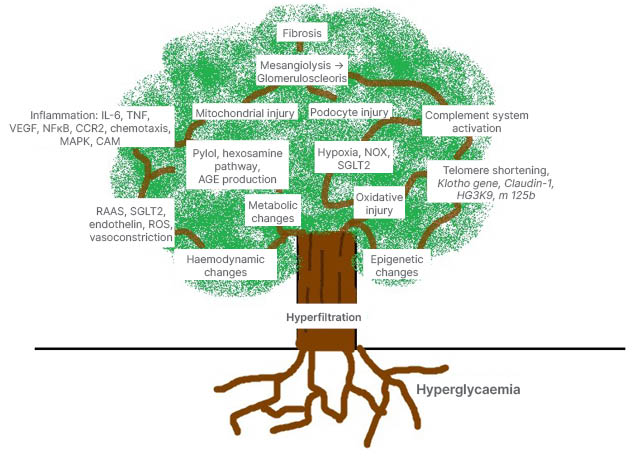
Figure 1: Hyperglycaemia lays the roots in the pathogenesis of diabetic kidney disease.
Hyperglycaemia leads to hyperfiltration, which is followed by metabolic, hormonal, haemodynamic, inflammatory and epigenetic changes. Oxidative stress and hypoxia play a vital role. All these precipitate to podocyte injury, mitochondrial distress, tissue death, glomerulosclerosis, and interstitial fibrosis.
AGE: advanced glycation end products; CAM: cell adhesion molecules; CCR2: C-C chemokine receptor 2; MAPK: mitogen activated protein kinase; NFкB: nuclear factor к B; NOX: nicotinamide adenine dinucleotide phosphate oxidases; RAAS: renin–angiotensin–aldosterone system; ROS: reactive oxygen species; SGLT2: sodium–glucose co-transporter 2; VEGF: vascular endothelial growth factor.
Hyperfiltration
Hyperglycaemia initiates its effects on the kidney by disturbing the osmotic forces. The higher blood glucose contents cause higher osmolality in the glomerular capillaries, resulting in higher glomerular pressures. This results in more outward forces increasing the glomerular filtration. Initially, this results in a falsely low to normal eGFR calculation based on more creatinine filtration with hyperglycaemia.6 This hyperfiltration is proposed to be multifactorial. Chemokines and enzymes like ornithine decarboxylase, which are produced in response to hyperglycaemia (noted in the kidneys of mice with hyperglycaemia), bring about renal enlargement with increased filtration surface area per nephron.7,8 Obesity, which can be integral to most patients with diabetes, can worsen renal enlargement and hyperglycaemia, promoting this initiation of renal injury.4,9 Weight loss in patients with DKD, through surgical9 or non-surgical methods in animal models, showed a significant reduction in the rate of decline in the eGFR.
Intraglomerular hypertension, which can be multifactorial, also predisposes to hyperfiltration. In many patients with diabetes, coexisting systemic hypertension,4,9 increased intrabdominal pressure due to obesity,10 and the increased osmotic pressures in the glomeruli end up contributing to this. Hyperglycaemia and protein-rich meals11 result in the production of various chemicals that can impair tubule-glomerular feedback, and increase local vasoconstrictors.
Another mechanism for hyperfiltration is the role of sodium–glucose co-transporters 2 (SGLT2) found in the proximal convoluted tubules. With supraphysiological levels of blood glucose, there is an upregulation of SGLT2, resulting in the maximal utilisation of these transporters. Maximal reabsorption in the PCT causes decrease tubular pressures, causing more filtration from the glomerular vessel.12 Also, with maximal sodium and glucose reabsorption, there is decreased distal sodium delivery activating the renin–angiotensin–aldosterone system (RAAS).
This hyper-flow state also leads to protein leak, which instigates alteration in the nephron structure, with resultant mesangial hypertrophy seen in the kidneys of patients with diabetes.
Measures taken to control hyperglycaemia have been proven to inhibit DKD progression, myocardial infarction, and mortality.13 Therefore, antihyperglycaemics have been the cornerstone in the management of DKD.
Haemodynamic Effects and Endothelial Injury
Hyperglycaemia and SGLT2-assisted decreased distal sodium delivery leads to activation of RAAS, producing efferent arteriolar vasoconstriction and afferent vasodilation, and amplifying intraglomerular hypertension. RAAS activation worsens systemic hypertension. Renin by itself has been noted to activate the mitogen activated protein kinase (MAPK) signalling pathway, while angiotensin II directly assists fibrosis by boosting transforming growth factor (TGF) production,14 vascular endothelial growth factor (VEGF), cell adhesion molecules, nuclear factor к B (NFкB) pathway, and toll-like receptors activation.15 TGF-β keeps a check between fibrosis and inflammation, and any imbalance in the concentration of TGF-β leads to abnormalities amongst them. TGF-β acts via SMAD3 and SMAD7. The former positively regulates non-coding RNA for inflammatory and fibrotic mediators, while the latter negatively affects it. Angiotensin II also stimulates production of reactive oxygen species (ROS) and inflammatory cellular attraction through activation of monocyte chemoattractant protein 1 (MCP-1), C-C chemokine receptor 2 (CCR2; in rat models),16 and co-stimulation of NK and T cells (in human subjects).17 In animal models and cultured kidney tissue, aldosterone was noted to facilitate fibroblast production by triggering TGF-β/SMAD2, fibronectin,18 stimulating platelet derived growth factor receptor plus EGFR via phosphoinositide 3-kinases/MAPK signalling,19 c-Jun N terminal kinase phosphorylation,20 and epithelial-mesenchymal transition.21 It also increases expression of plasminogen activator inhibitor-1, promoting hypercoagulable milieu.22 Angiotensin and hyperglycaemia prompt endothelin production, worsening vasoconstriction, inflammation, podocyte injury, nephrin shedding, and interstitial fibrosis.23
Metabolic Changes
Diabetes is associated with dysregulation of multiple metabolic pathways. Hyperglycaemia brings about activation of pathways including RHO/ROCK, hexosamine, pylol, advanced glycation end products (AGE), and protein kinase C (PKC), producing higher ROS, generating higher levels of MAPK, JAK signal transducers and activators of transcription, and NFкB,24 which are building blocks to inflammation and fibrosis. MAPK is linked to extracellular matrix production and podocyte injury.25 NFкB signals production of adhesion molecules and cytokines like macrophage chemoattractant protein MCP-1, IL-6, and tissue necrosis factor α.26 ROS also directly causes damage to cellular structures by oxidising various lipids, nucleic acids, and proteins. This lipid oxidation is aggravated with coexisting obesity and Type 2 diabetes, due to higher load of lipids.
Hypoxia and Oxidative Stress
Renal hypoxia is the inability of the oxygen supply to meet renal demands. This correlates directly with the blood supply, while the majority of demand depends on the metabolic activity in the tubules.
Hyperglycaemia can lead to oxidative stress and hypoxia in multiple ways. RAAS-mediated vasoconstriction causes ischaemic injury, hyperglycaemia causes hyperfiltration and tubular hypertrophy and overactivates SGLT2 channels, which depletes higher amounts of adenosine triphosphate and oxygen, leaving the nephron in a hypoxic state. An imbalance between antioxidants and ROS is a strong determinant for expediting tissue damage. Chronic hyperglycaemia also mediates overexpression and hyperactivity of nicotinamide adenine dinucleotide phosphate oxidases (NOX) 1, NOX2, NOX4, and NOX5.26,27 These produce higher ROS levels, causing Ras-related C3 botulinum toxin substrate 1 and VEGF-mediated foot process effacement, p53-driven apoptosis, protein kinase C, and A disintegrin and metalloprotease 17-mediated mesangial expansion, and uncoupling of nitric oxide synthase, causing vasocontraction.26,27 This hypoxia also produces hypoxia inducible factor which, under hyperglycaemic state, becomes unstable,28 and when chronically stimulated sparks a profibrotic effect.29 Ultimately, this becomes a vicious cycle of inflammation, vascular injury, and further hypoxia.28
Role of Inflammation and the Complement System
DKD has multiple intersecting pathways propagating its disease process. Inflammation plays a vital role in the pathogenesis of DKD. Diabetes sets in motion various inflammatory cascades via oxidative stress, AGE, obesity, ischaemia, and damaged cells,30 producing inflammatory molecules like NFкB, NLR family pyrin domain containing 3 (NLRP3)-linked caspases,31 IL-1B, IL-6, and IL-18. This increase in AGE has been shown to be linked directly with increased expression of NLRP3-related proteins, which have been postulated to be mediators of chronic kidney disease,31 with a role in activation of mesangial cells. NLRP3-related proteins are found in macrophages and inflammasomes and have been linked to multiple inflammatory disorders. In various mice models, attenuation of NLPR3 helped reduce chronic kidney disease (CKD) progression in a dose-dependent manner. Neutrophil and macrophage infiltration, lipoprotein oxidisation, and immune complex deposition can also occur.6 With this ongoing inflammation there is increased production and deposition of amyloid A protein, which can also be used as a marker of disease progression.32 CCR2 signalling distorts actin cytoskeleton and nephrin stability, damaging podocytes.33 Hyperglycaemia-mediated promotion of elective cell simulated adhesion molecules cause tight junction abnormalities, resulting in proteinuria.33
Furthermore, the complement system activation has a huge impact on DKD progression. DKD progression has been linked to complement activation through mannose-binding lectins and ficolin-associated activation of the lectin pathway in the complement cascade. Hyperglycaemia leads to higher levels of glycan and galactosamine-bound substances that are recognised by these receptors.34
Genetic Modification
Hyperglycaemia and its effects cause DNA damage, and display effects of ageing in patients with diabetes by causing chromosomal telomere shortening,35 resulting in proteinuria and DKD progression.36 DNA damage activates various kinases, including ataxia-telangiectasia mutated and Rad3-related, followed by activation of p51 and p21. This inhibits cyclin-dependent kinase 2, impeding phosphorylation of retinoblastoma protein, which is essential for E2F transcription factor mediated DNA transcription. This inhibition of E2F transcription factor leads to permanent arrest of the cell cycle.30
Epigenetic changes constitute changes in genome function without change in DNA sequences. These are directly linked to lifestyle-directed exposures and individual encounters. In mice with diabetes, epigenetic changes have been noted as early as after 5 weeks of hyperglycaemia. DNA methylation, histone modification, and non-coding RNA are the main epigenetic changes.37 Histone acetylation of HG3K9 causes increased expression of AGT, a component of RAAS system,38 and hypomethylation of CpG islands (which are portions of some regulatory regions of genome) causes changes in the Claudin-1 gene, resulting in more proteinuria.39 H3K4me1 methylation promotes NFкB, non-coding RNA miRNA125b, prompting IL-6 and MCP-1 expression and miRNA-192-led TGF-β activity, plus collagen accumulation.40 Hyperglycaemia also provokes activity of HDAC4, which deacetylates signal transducers and activators of transcription 1, inhibiting autophagic properties in podocytes.41 Numerous other epigenetic changes play a role in promoting oxidative stress: e.g., hypomethylation of CpG or hyperacetylation of H3 histone precipitates p66Shc promotion, and lysine-specific histone demethylase 1 histone dimethyltransferase-driven inhibition of SOD2.42
Hypermethylation of klotho gene in both patients and mice with diabetes results in lower levels of klotho protein.43 This decreases the favourable effects of klotho protein against ageing, oxidative stress calcification, and antifibrotic effect.44,30
Kidney Changes
The kidneys in a diabetic environment undergo many changes, from initial renal enlargement to vasoconstriction, endothelial and tubular cell injury, to eventual renal fibrosis. The authors describe certain structural changes and their places in the pathogenesis of diabetic kidney below.
Glomerular Changes
The earliest changes in DKD are due to hyperfiltration in the glomerulus, causing thickening and stiffness of the glomerular basement membrane from sheer pressure45 and deposition of extracellular matrix.46 Secondly, mesangial expansion occurs due to leakage of protein, inflammation, and ongoing damaged tissue collection. This further disturbs the precision of glomerular filtration. Mesangiolysis leads to accumulation of matrix and cellular debris, which forms nodular structures named Kimmelstiel and Wilson nodules.47 Additional mesangial destruction ends up in widespread glomerulosclerosis. Vascular changes with thickening of vessel walls and hyalinosis are also classic for DKD.
Podocyte Injury
Podocytes are the building blocks of the renal system. They are the prime managers of the filtration system. Podocyte injury has been shown to mimic diabetic changes even in absence of hyperglycaemia,48 which indicates that podocyte injury is the key in development of DKD. Hyperglycaemia, oxidative stress, and inflammation leads to podocyte effacement, actin rearrangement, increased tight junction, slit diaphragm abnormalities, and apoptosis. In mice models, activation of mammalian target of rapamycin complex 1,49 dynamin-related protein in mitochondria, nicotinamide adenine dinucleotide phosphate oxidase, and AMP-activated protein kinases50 are responsible for these changes.
Cellular and Mitochondrial Injury
The renal tubules, given their high metabolic demand, are rich in mitochondria. Patients with diabetes have been noted to have mitochondrial abnormalities, including mitochondrial fragmentation, decreased adenosine triphosphate, increased mitochondrial permeability, and mitochondrial uncoupling as early as 4 weeks after hyperglycaemia. Peroxisome proliferator-activated receptor-gamma coactivator-1α is amongst the prime regulators of mitochondrial synthesis, and its expression is altered in DKD.51 The electron transport chain subunits in the mitochondria are also directly damaged by the oxidative stress that occurs with hyperglycaemia, leading to worsening mitochondrial metabolic functions51 via DNA damage and decreased activity of glyceraldehyde 3-phosphate dehydrogenase. This leads to shifts in the glycolytic pathway to pylol and hexosamine pathways. Resultant oxidative stress causes decrease in AMP-activated protein kinase activity, leading to NFкB-mediated inflammation.6
Fibrosis
Unfortunately, all of the above mechanisms merge together and result ultimately in fibrosis and atrophic kidney tissue. The degree of tubulointerstitial fibrosis can even supersede glomerular lesions in determining renal function.52 Local myofibroblasts, bone marrow-derived fibrocytes, and epithelial to mesenchymal transition as a response to the chemokines have been noted to produce this effect. Figure 2 shows some of these effects.
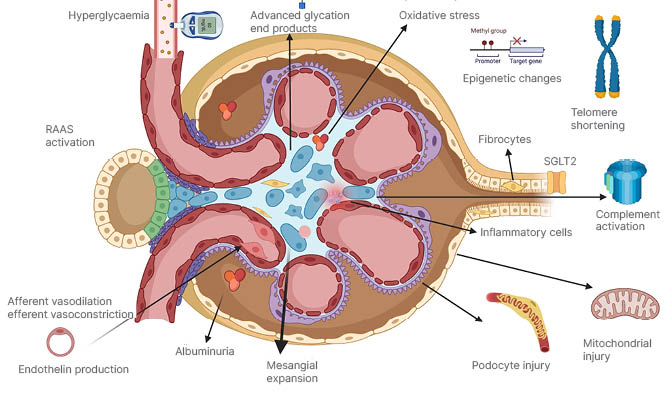
Figure 2: Depicts the glomerulus and tubular cells with some changes that happen in diabetes.
RAAS: renin–angiotensin–aldosterone system; SGLT2: sodium–glucose co-transporter 2.
Target Therapies
Most of the pathway mediators that have been identified are potential targets to slow down DKD progression. Many of them have been tried and even more are in the pipeline. Table 1 summarises the ones mentioned below.
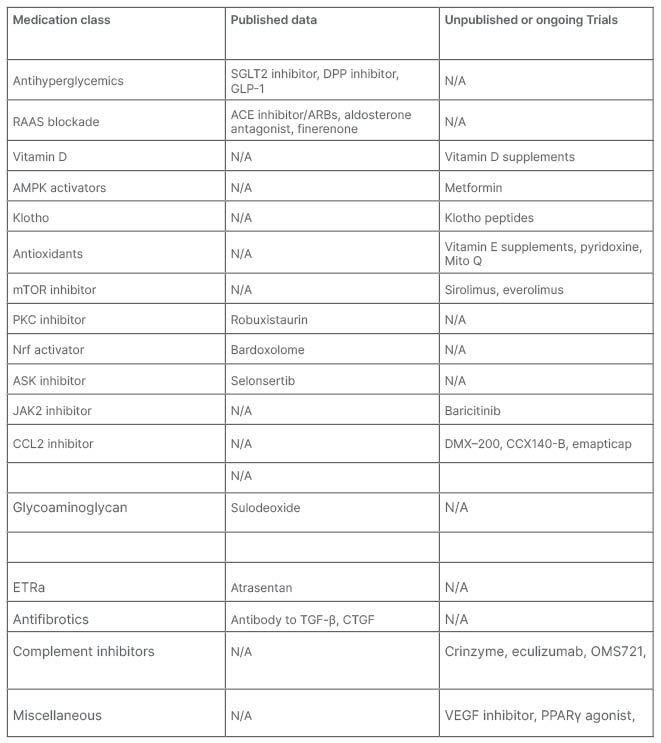
Table 1: Pathway mediators that have been identified as potential targets to slow down the progression of diabetic kidney disease.
ACE: angiotensin-converting enzyme inhibitor; AMPK: AMP-activated protein kinase; ARB: angiotensin receptor blockers; CCL2: chemokine (C-C motif) ligand 2; DPP: dipeptidyl peptidase; CTGF: connective tissue growth factor; ETRa: endothelin receptor antagonist; GLP-1: glucagon-like peptide-1; mTOR: mammalian target of rapamycin; N/A: not applicable; PKC: protein kinase C; RAAS: renin–angiotensin–aldosterone system; SGLT2: sodium–glucose co-transporter 2; VAM: vinyl acetate monomer; VEGF; vascular endothelial growth factor.
RAAS Blockade
angiotensin-converting enzyme inhibitors/angiotensin receptor blockers and aldosterone antagonists have been the mainstay for management of DKD for decades. Emerging evidence of nonsteroidal mineralocorticoid receptor antagonist, finerenone, is an exciting prospect as well.
Vitamin D supplementation has also been noted to decrease the levels of renin.53
Antihyperglycaemics
Hyperglycaemia is probably the instigator for all the downstream effects leading to DKD. Multiple medications have been in use to control diabetes. Particular attention has been received by some of the antihyperglycaemics for their renoprotective and cardioprotective effects than just lowering blood glucose.
• Glucagon-like peptide-1 analogues showed benefit in trials like SUSTAIN 6, REWIND, AWARD7, and AMOLITUDE-O. The FLOW trial is still ongoing.
• Dipeptidyl peptidase inhibitors showed benefit against albuminuria in MARLINA and CARMELINA trials.
• SGLT2 inhibitors have been the new breakthrough in DKD management in the last decade. Multiple studies have shown improvement in cardiac mortality, proteinuria, and prohibiting disease progression. CREDENCE and DAPA CKD have proven their utility in DKD. EMPA Kidney is the most recent trial investigating this, and is currently ongoing.
AMPK Activators
Metformin has been shown to activate AMPK levels, decreasing ROS production, and slowing DKD.54
Klotho Peptides
Klotho peptides is an anti-ageing protein which is found to have decreased levels in patients with diabetes. Its supplements have been hypothesised to help slow down the progression of DKD.55
Antioxidants
Oxidative stress and ROS are notorious for their injurious properties throughout the body. In DKD, multiple antioxidants have been shown to slow down the process. Vitamin E supplements,56 pyridoxamine,57 and mitoQ (in mice)58 have been investigated.
mTOR Inhibitors
Rapamycin has been shown to slow down DKD progression in animal models; however, it has yet to go through human trials given its side effect profile.
Anti-inflammatory
Many inflammatory mediators have been targeted in hopes of slowing down the disease process. Some of them are mentioned below:
• Ruboxistaurin, a protein kinase C inhibitor, was noted to decrease albuminuria.
• Bardoxolone is an Nrf activator, with ongoing trials MERLIN and AYAME.
• Selonsertib is an ASK inhibitor undergoing MOSAIC trial.
• Baricitinib a JAK2 inhibitor, showed improvement in Phase 2 trials.
• Chemokine (C-C motif) ligand 2 or MCP-1 inhibitors DMX–200 and CCX140-B59 and emapticap improves albuminuria.
• VEGF antibody trial undergoing recruitment.
• Sulodeoxide, a glycoaminoglycan composed of low molecular weight heparin, showed a decrease in albuminuria.60
• Pentoxifylline, a phosphodiesterase inhibitor, showed hopeful results in the PERIDIAN trial in 2015, with a larger trial ongoing.
• Atrasentan, an endothelin A receptor antagonist, showed promise in renal disease prevention (SONAR trial).
• Under trial: nicotinamide adenine dinucleotide phosphate antagonists, PPAR-Γ agonist, vascular adhesion molecules inhibitor.
• Anti–NLRP3 in rat models: Phenethyl isothiocyanate,61 and naringin.62
Antifibrotics
Pirfenidone, an antibody against TGF-β and connective tissue growth factor was terminated due to side effects, but did reduce albuminuria. Some interest remains in minocycline, while N acetylcysteine did not show any positive outcomes.
Complement Inhibitors
Multiple complement inhibitors are being targeted in trials: C3a receptor and C5a receptor inhibitors, eculizumab, C-1 inhibitor (crinzyme) inhibits mannose-binding lectin due to structural similarity. OMS721 is an antibody against mannose-binding lectin associated serine protease 2. Anti-CD59 inhibits MAC.63
NOVEL APPROACHES TO EARLY DETECTION OF DIABETIC KIDNEY DISEASE
Despite their limitations, the authors have relied on creatinine-based eGFR calculation, urine albumin, and creatinine excretion ratios as tools of detection and progression of DKD. Combining cystatin C and creatinine-based eGFR calculation has improved estimation of kidney function, but still does not help in earlier detection of the disease. In recent years, due to advancements in the fields of metabolomics, transcriptomics, and bioinformatic analysis, it has been possible to detect earlier changes, given better understanding of the components in the pathophysiology of DKD, which has become an alternative to estimate kidney function due to inaccuracies of eGFR. Neutrophil gelatinase-associated lipocalin and kidney injury molecule-1 showed a role in detecting glomerular hyperfiltration.64 Development of DKD can be predicted with urinary levels of N-acetyl-β-D-glucosaminidase and 8-oxodG.64 Urinary levels of pentosidine65 and serum levels of tissue necrosis factor receptors66 were predictive of progression of albuminuria and DKD, respectively. DNA 65 (a panel with fragments of collagen 1)67 and CKD 273 (a set of 273 urinary peptides)68 helped to distinguish and detect DKD earlier. Other metabolomic studies have shown various changes in metabolite levels that can indicate DKD earlier. These metabolites are from metabolic pathways involving fatty acids, phospholipid oxidation, urea cycle, and amino acid metabolism, to name a few.69 Some of the indicative metabolites that are increased in DKD include aspartic acid, citruline, symmetric dimethylargine, and kynurenine.69 Levels of uremic solutes, fatty acids, amino acids, histidine, butenoylcarnitines, acyl carnitines, and branched chain fatty acids were able to predict progression of DKD years in advance.69 A recent comparison of 232 patients with 100 healthy individuals showed that certain metabolomic markers, like dodecanoylcarnitines, triglylcarnitine, and isovalerylcarnitine, strongly predicted albumin creatinine ratio.70
CONCLUSION
We have come a long way in discovering the mechanics behind the destructive pathology of DKD, but there still remains a lot of work to be done. The unpredictability in the disease course of DKD, due to its nonlinear pattern of eGFR decline,71,72 indicates the variety of factors affecting its pathogenesis. We now have a better picture of how high glucose levels involve haemodynamic changes, changes in hormone production, metabolic pathway alteration, oxidative stress, and worsening inflammation. It is clear how diabetes not only involves the podocytes and the glomeruli, but also damages the tubules. The damage caused is so widespread that it involves changes on the cellular level, including mitochondrial injury and epigenetic changes. Understanding DKD at a molecular level helps with the ability to tailor management, as we try to prevent ESKD or complications of DKD.