
Meeting Summary
This symposium took place during the International Conference on Malignant Lymphoma (ICML) in Lugano, Switzerland, June 2019, and focussed on scientific aspects associated with development and approval of rituximab biosimilars in lymphoma. The symposium began with an overview presented by Dr Cornes detailing the urgent economic need for biosimilars to improve access to these biologic treatments in oncology and other therapy areas. Prof Schellekens, author of the first paper on biosimilars in 2002, discussed how regulatory strategies for biosimilars were shaped, and how these have evolved in the intervening years. Today, the emphasis of biosimilar development is placed on extensive analytical testing to demonstrate a match with the reference medicine at a fundamental level. Clinical testing plays a confirmatory role, removing any residual uncertainty regarding potential clinical differences between biosimilar and reference medicine. Dr Schiestl presented further detail on analytical perspectives on biosimilars. Development of biosimilars is complicated by the inherent variability of biological synthesis techniques employed in the manufacture of biologics. This variability is further increased by ongoing changes to manufacturing processes, which can result in changes in biological activity. Consistent quality is therefore a cornerstone of biosimilar development. Prof Jurczak provided a comprehensive overview of the factors that must be considered during clinical development of a biosimilar. Clinical trials for biosimilars have a confirmatory role in the development process, rather than the pivotal role played by clinical trials for reference medicines. Therefore, these trials have markedly different objectives compared with reference clinical trials, resulting in differences in the chosen endpoints. In biosimilar trials, response endpoints, which provide rapid and sensitive assessments of equivalence, are preferred to survival endpoints, which require large and lengthy trials for adequate evaluation. Prof Jurczak illustrated this using data from the Phase III clinical trials of the Sandoz rituximab biosimilar. In this trial, Sandoz rituximab demonstrated an equivalent response rate to reference rituximab. Increasing economic pressure on healthcare systems means that biosimilars are likely to play an increasing role in the treatment of cancer in coming years, requiring clinicians to increase their familiarity with these important medicines.
Introduction to Biologics and Biosimilars
Doctor Paul Cornes
The World Health Organization (WHO) states that access to essential medicines is part of the right to health.1 However, patterns of disease are changing, with noncommunicable diseases (NCD) becoming more common, now being responsible for almost 70% of all deaths worldwide.2,3 To adapt to these changes in patterns of disease and control NCD, multiple innovations are required across domains of prevention, early diagnosis, and treatment. In particular, innovation in medicines is required – better treatment means more effective medicines for more diseases.3 Of all NCD, cancer is a key threat, and this is universal to all countries globally.3,4 Substantial innovation in cancer medicines has been achieved in recent years, and at the current rate >100 new cancer drugs could be available by 2020, giving a total of 200 cancer medicines.5-8 Furthermore, >700 molecules were in late-stage development in 2017, an increase of >60% from a decade ago; almost 90% of these drugs are targeted treatments, including biologics.9
Biologic drugs are transforming treatment of other hard-to-treat diseases beyond cancer, including multiple sclerosis, heart disease, asthma, and inflammatory bowel disease,10 because these targeted therapies offer improved effectiveness compared with past generations of small-molecule medicines.11 However, biologics attract high costs, and therefore represent a good target for cost-control initiatives. Speciality medicines, including biologics, represent a large proportion of the overall medicines budget, ranging from 18–35% in South Korea, Japan, Canada, and Australia, to 35% in the USA and 35–45% in the five largest European markets (2016).12
Issues of medicine affordability are becoming evident in many therapy areas, including but not limited to oncology.9 The median annual cost per life-year gained of a new cancer drug launched in 2014 exceeded $200,000,13 and novel checkpoint inhibitors all carry list prices of more than $12,000 a month.14 The best-selling anti-inflammatory biologic Humira (adalimumab) costs $38,000/year in the USA,15 which equates to more than half the median income of an American household. Overall, total global prescription medicine sales are expected to be $1.2 trillion in 2024, and are increasing by 6–7% year-on-year.16
These high costs inevitably result in an impact on access to drugs. Evidence suggests that there are serious gaps in the availability of basic chemotherapeutic and biologic medicines in many Central and Eastern European countries.17 In addition, a strong correlation exists between the wealth of a country and the number of patients receiving biologics,18 with only patients in the USA, Germany, and the UK having access to >40 of the 54 oncology medicines initially launched between 2013 and 2017.19 This exemplifies the stark reality; globally, cancer care is not affordable for most patients and many governments,20 but the rise in cancer and other NCD cannot be solved without innovation in medical treatments. Therefore, healthcare budgets need to change to bear the costs of innovation; however, this can only be achieved by making savings elsewhere, which in turn must not compromise care. One potential route to such savings is the use of generic and biosimilar medicines to replace originator medicines. Between 2018 and 2024, it is predicted that $251 billion of currently patent-protected sales could be replaced by more affordable follow-on versions.16
Seven of the top ten selling medicines in 2017 were biologics, three for treatment of inflammatory disease and four for treatment of cancers.21 All of these biologics have, or will soon have, biosimilar brand competition. In 2017, sales of these seven biologics totalled more than $63 billion.21 With an estimated 33% price saving offered by biosimilars, >$20 billion a year could be saved and used to sustain global healthcare. Previously, experts have discussed how the demand for biologics is putting pressure on healthcare budgets, suggesting that the introduction of biosimilars could potentially greatly reduce costs and increase access to treatment.22 However, the manufacturing and development of biologics and biosimilars is complex. This was the focus of the symposium and will be discussed in subsequent sections of this report.
Biologics and Biosimilars: The Regulatory Perspective
Professor Huub Schellekens
The first article describing the development of ‘off-patent biotech products’, which would come to be known as biosimilars, was published in 2002 by Schellekens and Ryff.23 The publication outlined the need for a new regulatory pathway for this new class of products to augment the existing pathway used for generic medicines. While generics are copies of small molecules that are easy to synthesise chemically, biosimilars are new versions of biologics, which are relatively large and complex proteins manufactured in living systems. The enormous complexity of the cell machinery used in the manufacturing process means that biologic manufacture cannot be rigidly controlled, and a certain degree of variability is present in all biologics, both within and between batches.24 This variability results in challenges for biosimilar manufacturers, in that they must manufacture a product to a moving target. A further complication is that the proprietary technology used to develop the original molecule is unknown to the biosimilar manufacturer. Therefore, an iterative process is required to gradually guide the fingerprint of the biosimilar towards that of the reference medicine.25
The 2002 publication by Schellekens and Ryff23 also suggested that extensive clinical data would be needed to show the efficacy and safety of a proposed biosimilar, to an extent similar to the reference biologic. In 1998, a formulation change for an epoetin molecule had resulted in the development of cross-reacting anti-epoetin antibodies leading to pure red cell aplasia.26 This in turn led to an assumption that the clinical performance of biologics could be significantly altered by even small changes in the manufacturing process.
The approach to biosimilars has advanced considerably since 2002. Precision analytical tools are used to provide an extensive physicochemical and biological characterisation of the biosimilar, conclusively demonstrating high similarity to the reference medicine (Figure 1).27 Once these analytical comparisons are complete, the regulatory pathway recommends clinical comparison of pharmacokinetics (PK) and pharmacodynamics (PD), followed by a single Phase III clinical study, which is considered confirmatory to resolve any potential uncertainty regarding the similarity of the biosimilar candidate and the reference medicine.27
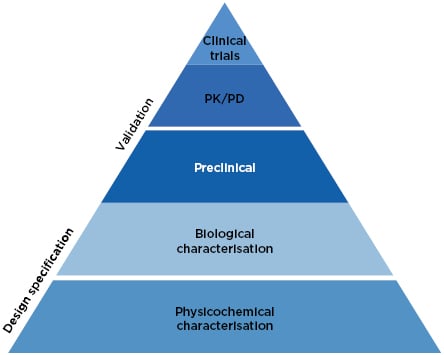
Figure 1: The current approach to biosimilar development.27
PD: pharmacodynamics; PK: pharmacokinetics.
Since 2006, the date of the first biosimilar approval in Europe, we have also gained knowledge on the safety of biologics, and the idea that ‘even minor differences may affect the safety and/or efficacy of biologics’ is no longer a consensus opinion. Manufacturing changes for biologics occur at an average rate of 1.8 per year.28 For example, to date, 29 high-risk or moderate-risk process changes have been observed for reference rituximab.29 These changes have not resulted in withdrawal of the products or alterations in their labelling, indicating that the observed changes were predicted by health authorities to not result in an altered clinical profile.30 Furthermore, no unexpected safety issue has been reported for currently available biosimilars on the European market, including more than 58 medicines and over 700 million patient-days’ exposure.31 As noted by Lamanna et al.,32 “biopharmaceutical variability resulting in clinical differences is extremely rare with only a single verifiable case resulting in adverse events in over 35 years and over 260 products.”
Biologics and Biosimilars: The Analytical Perspective
Doctor Martin Schiestl
Biosimilars are precisely engineered to match an existing reference biologic medicine in all relevant attributes. Based on guidance published by regional and national health authorities, key requirements for biosimilarity are matching structure and function; similar PK/PD, clinical efficacy and safety; and the same presentation, dose (strength), and method of administration. Biosimilars must have certain relationships with their reference medicines in terms of structure. The amino acid sequence must be identical, while secondary, tertiary, and quaternary folding patterns must be indistinguishable across multiple redundant methods of analysis. Owing to biosynthesis in living cells, post-translational modifications such as glycosylation and sialylation demonstrate a certain degree of variability for biologic medicines, and each batch of a given biologic can be differentiated from other batches using sensitive analytical methods.30
Manufacturing changes may result in more profound shifts in attributes, which in some cases may be associated with changes in biological function. For example, the antibody-dependent cellular cytotoxicity activity of reference rituximab was noted to undergo a noticeable shift following a change in the manufacturing process in batches expiring after mid-2010.30 However, this variability is tightly controlled within acceptable limits to ensure that such shifts have no relevant clinical impact. Moderate and major manufacturing changes also require pre-approval by the health authorities who grant such a change only if the risk for any clinical impact is sufficiently low. To exclude relevant clinical differences of a biosimilar to its reference medicine, the biosimilar must match the reference medicine as closely as possible in terms of physicochemical structure, with particular focus on attributes known to have the largest potential impact on clinical performance.25 This close similarity at a molecular level can then be confirmed using in vivo biological testing. For example, Sandoz rituximab was shown to have biological activity that fell within the range of reference rituximab across the key effector functionalities associated with the central mechanism of action.33
Consistent manufacturing of biosimilars is ensured through control of the manufacturing process, including control of raw material, process design, in-process testing, and control of process parameters, release testing of harvest, drug substance, and final dosage form. The quality system is governed by Quality Assurance functions, compliance with Good Manufacturing Practices (GMP) and regular inspections by health authorities. Dr Schiestl concluded that consistent quality is a key regulatory requirement for biosimilars, and analytical capabilities and quality systems are continuously improving, allowing for robustness in ensuring consistent quality.32,34 A biosimilar medicine and its reference medicine are expected to have the same safety and efficacy profile,10,35 as confirmed by 13 years of clinical experience with biosimilars in the European Union (EU) as of 2019.36
Biologics and Biosimilars: The Clinician’s Perspective
Doctor Paul Cornes and Professor Wojciech Jurczak
The Biosimilars Forum Survey, conducted in 2015–2016, aimed to assess physicians’ knowledge of biosimilars.37 When asked ‘Do you believe biosimilars will be safe and appropriate for use both in naïve and pre-exposed patients?’, <50% of respondents were in agreement. Prof Pekka Kurki of the Finnish Medicines Agency has suggested that the root cause of this uncertainty is a discrepancy between the needs of regulators, who assess the totality of evidence, and clinicians, who focus only on clinical testing.38 Attitudes towards biosimilars are improving however. An analysis of publications mentioning biosimilars between 2004 and 2015 found exclusively sceptical language in 2004, which had changed to be approximately 50% positive and 40% neutral by 2015.39
The common desire to see clinical data is learnt from deep familiarity with the development process for innovator drugs, for which clinical trials are indisputably the optimal way to confirm efficacy.40 In biosimilar development, however, the overall goal of the clinical programme is to demonstrate equivalence of the biosimilar to the reference medicine. The corresponding statement is that the clinical programme aims to detect any difference between the medicines, should it be present. Therefore, the indication chosen for a clinical trial should be sensitive to detect any potential differences, rather than the most frequently prescribed indication for the drug.41 Similarly, the appropriate endpoints could be very different to ‘traditional’ Phase III endpoints; PD endpoints are likely to be more sensitive than clinical endpoints.42 In clinical trials of reference trastuzumab, for example, the PD endpoint pathologic complete response (CR) was shown to be four times more sensitive than overall survival, and achieved in one tenth of the time.43 Overall, the design features differ in studies for new biologics and for biosimilars, as summarised in Table 1.44,45
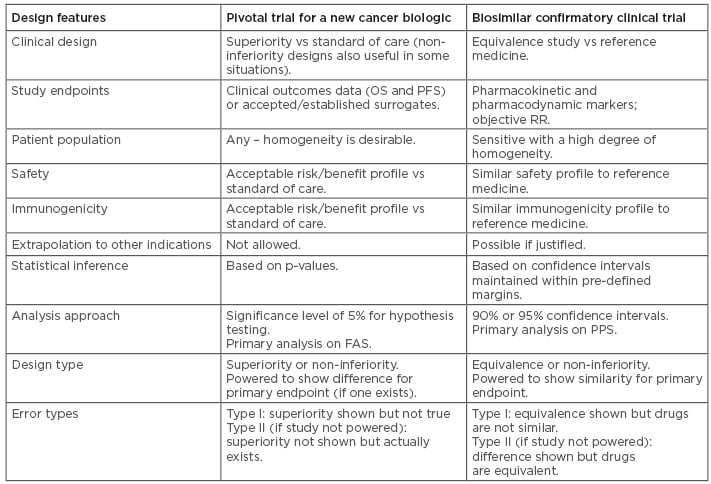
Table 1: Comparisons between study designs for a new biologic and for a biosimilar.44,45
FAS: full analysis set; OS: overall survival; PFS: progression-free survival; PPS: per-protocol set; RR: response rate.
An equivalence trial is designed to provide statistical evidence that there are no clinically meaningful differences between the biosimilar and its reference medicine.46 Therefore, the critical first step in biosimilar trial design is to predict the effect of the reference medicine on the chosen endpoint. Then, it is necessary to determine how large a difference could be observed between the effect of the reference medicine and the proposed biosimilar before it would be regarded as non-equivalent; this difference is known as the equivalence margin.46 Generally, trials are designed to test whether the true difference lies within the equivalence margin with 90% or 95% confidence. If this is the case, then the treatments can be considered equivalent.
Trial designs must also take account of whether to use the risk difference or risk ratio to compare treatments. ‘Risk difference’ measures the absolute difference between two treatments, where equivalence is denoted by a difference of 0. Conversely, ‘risk ratio’ measures the relative difference between two treatments, where equivalence is denoted by a difference of 1.47 Importantly, these points are generally agreed with regulatory authorities before a trial begins.
Survival endpoints (progression-free survival [PFS], overall survival [OS]) are included in almost all trials for originator oncology drugs. However, these endpoints have limitations that make them less suitable in certain situations, including in the development of biosimilars.44 For example, survival endpoints require longer follow-up times and larger sample sizes for reliable measurement compared with response-based endpoints.48 Importantly, survival endpoints do not directly measure the antitumour activity of a drug, and may be influenced by factors not attributable to differences between the biosimilar and reference biologic, including tumour burden, performance status, underlying conditions, and previous and subsequent treatments.44,49 Therefore, regulatory authorities including the European Medicines Agency (EMA) recommend that endpoints such as response rate may be more sensitive than PFS or OS for detecting differences between a biosimilar and reference medicine, and can be used as surrogates for survival in biosimilar trials.44
The clinical development programme for Sandoz rituximab included studies of PK/PD, efficacy and safety from two indications, follicular lymphoma (FL) and rheumatoid arthritis. FL was chosen as an appropriate indication for a Phase III confirmatory clinical trial, as this indication resulted in the largest effect size for rituximab when added to standard chemotherapy, and is the most homogeneous of the approved oncology indications for rituximab.50 The ASSIST-FL study was a randomised, Phase III trial of efficacy, safety, and PK of Sandoz rituximab versus EU-sourced reference rituximab.50 Overall response rate (CR or partial response [PR]) was chosen as the most appropriate endpoint for the trial, as to confirm similarity with an overall survival endpoint would have required several thousand patients. Given that large clinical trials (>1,000 patients) have a mean cost of $77 million,51 this would have been prohibitive.
The study included patients with previously untreated, advanced-stage FL (Ann Arbor stage III/IV, WHO histological grade 1-3a) who were randomised to receive either Sandoz rituximab 375 mg/m2 + cyclophosphamide, vincristine, and prednisone (CVP) chemotherapy (n=314) or EU-reference rituximab 375 mg/m2 + CVP (n=315) for eight 3-weekly treatment cycles, known as the induction phase or combination treatment period. Tumour assessment was performed at end of Cycles 4 and 8. During the 2-year maintenance period, responders received Sandoz rituximab (n=254) or EU-sourced reference rituximab (n=252) every 3 months. The primary endpoint was met: equivalence in overall response rate was demonstrated, with a difference between treatments of -0.40% and a 95% CI of -5.94 to 5.14. This was entirely contained within the predefined equivalence interval of -12 to 12%, as was the 90% CI (-5.10 to 4.30%) (Figure 2).50
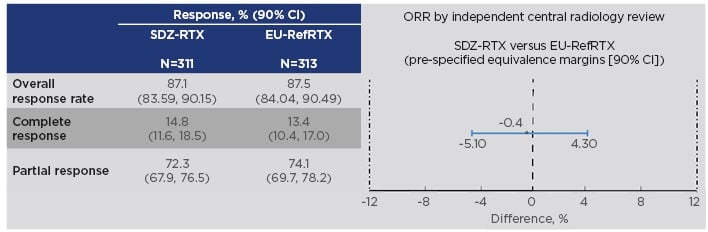
Figure 2: Results on the primary endpoint in the ASSIST-FL trial of Sandoz rituximab.50,52
CI: confidence interval; EU-RefRTX: EU-sourced reference rituximab; ORR: overall response rate; SDZ-RTX: Sandoz rituximab.
In ASSIST-FL, PFS and OS were unpowered, descriptive secondary endpoints.52 CR after 30 months (CR30) is considered a surrogate for PFS, as correlation between these two outcome measures has previously been established.53 CR rates (based on investigator assessment) were similar between treatments at all time points, including 33 months.52 Safety profiles of Sandoz rituximab and reference rituximab were similar when combined with CVP in the combination phase, or alone in the maintenance phase.50 Incidences of adverse events (AE), serious AE, and AE leading to discontinuations and deaths were comparable. Most AE were mild or moderate in severity.
Conclusions
Doctor Paul Cornes
The implementation of biosimilars is a central policy imperative for the EU.54 At a stakeholder event on biosimilar medicines held by the European Commission in Brussels in September 2018,55 the consensus stated that there are no outstanding serious medical issues regarding biosimilars, that they can be considered as safe and of the same quality as reference biologics, and that they may be associated with significant cost savings.35,54 Stakeholders also agreed that biosimilars can be used in indications that are approved for the reference medicine but not studied for the biosimilar, enabling pharmacies to stock only one brand,56 and that brands can be switched safely,57 possibly as part of the annual tender process. In a decade of use (with >58 biosimilars now approved and >700 million patient-days’ exposure) EMA-approved biosimilars have all maintained approval without showing a different risk or benefit profile to the reference medicine. Within 12 months of launch, biosimilar rituximab infusions overtook the reference biologic in the EU5 nations.58 Ongoing increases in biosimilar uptake are likely to lead to considerable healthcare savings,16 broadening access to biologics for patients in wealthy as well as lower income countries, ultimately allowing for increased sustainability of care and an increased budget for development of innovative treatments.





