
Meeting Summary
Several rare haematological diseases are linked to bone marrow failure (BMF). This symposium provided the latest scientific insights into the different pathophysiological mechanisms and clinical advances in the management of these conditions, with a specific focus on the clinical management of patients with paroxysmal nocturnal haemoglobinuria (PNH) in the context of aplastic anaemia (AA), and the pathophysiology, consequences, and identification of PNH in the context of BMF.
Prof Gérard Socié chaired the symposium and overviewed BMF. Dr Austin Kulasekararaj gave a presentation on new paradigms in BMF, followed by Prof Gérard Socié, who reviewed the diagnosis and management of AA. Dr Alexander Röth then discussed the diagnosis and management of PNH in the context of BMF. The symposium was concluded by a short question and answer session.
Overview of Bone Marrow Failure
Professor Gérard Socié
Several different diseases overlap within BMF syndromes and share the propensity to develop into myelodysplastic syndromes (MDS) and acute myeloid leukaemia (AML) (Figure 1).1 For any BMF or AA, especially in children or young adults, a congenital disorder should be ruled out during diagnosis. Patients with congenital BMF will not respond to immunosuppressive therapy (IST).
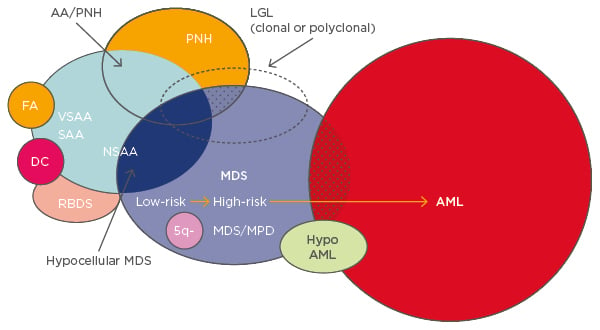
Figure 1: Overlapping entities in bone marrow failure syndromes.1
AA: aplastic anaemia; PNH: paroxysmal nocturnal haemoglobinuria; FA: Fanconi anaemia; VSAA: very severe aplastic anaemia; SAA: severe aplastic anaemia; NSAA: non-severe aplastic anaemia; DC: dyskeratosis congenita; RBDS: ribosomal dysgenesis syndrome; 5q-: q arm of chromosome 5 deletion; MDS: myelodysplastic syndrome; MPD: myeloproliferative disorder; LGL: large granular lymphocytic leukaemia; AML: acute myeloid leukaemia.
Anaemia is the most common clinical sign of BMF, others include neutropenia, monocytopenia, and thrombocytopenia. Blood analysis is important for differential diagnosis to detect abnormalities in neutrophils, platelets, blast cells, and other cells. During bone marrow analysis mast cells and plasma cells are frequently detected in patients with severe AA, increased erythroblasts frequently detected in PNH, and dyserythropoiesis in both.
Bone marrow aspirate and trephine biopsy are fundamental to a differential diagnosis. However, it is important to consider that ageing is also associated with reduced bone marrow cellularity.2 Theoretically, distinguishing between AA and hypoplastic MDS is relatively simple, as dysplastic neutrophils and, in particular, dysplastic megakaryocytes are key features of hypoplastic MDS but not of AA.3 CD34+ cells may also be used. Cytogenetic analysis plays a key role in diagnosis, as hypocellular bone marrow can lead to insufficient cellular metaphase. Fluorescence in situ hybridisation for chromosomes 5, 8, and 7 should be considered for differential diagnosis. However, an abnormal cytogenetic clone does not imply a diagnosis of MDS, particularly as trisomy 8 is frequently detected in patients who are otherwise typical for AA. Flow cytometry is a key diagnostic tool for PNH and will be discussed in depth later. Other diagnostic tools may include screening for viral hepatitis or autoimmune disease (although cases linked to these diseases are rare) and vitamin B12 dosing.
Two rare diseases to consider in children, and adolescents and young adults, are Fanconi anaemia, which can easily be discounted using a chromosomal breakage test, and dyskeratosis congenita. Over recent years, the number of patients with a diagnosis of dyskeratosis congenita has increased, even though the disease development presents a wide spectrum of clinical features. A family history of pulmonary fibrosis, unknown pulmonary complications, or non-alcohol-related cirrhosis should be investigated, and dyskeratosis congenita has a frequent association with mutations of the telomerase RNA compartment (TERC) or telomerase reverse transcriptase (TERT). Despite the variety of tests available, in practice differential diagnosis between MDS and severe AA/PNH is not necessarily easy.
New Paradigms in Bone Marrow Failure
Doctor Austin Kulasekararaj
AA is an immune-mediated non-malignant disease, and clonal haematopoiesis in AA is linked to the evolution of disorders such as PNH and MDS/AML, which are associated with acquired genetic abnormalities.3 Recently, studies have identified inherited GATA2 mutations in patients presenting with AA, MDS, and also paediatric neutropenia, but invariably with severe monocytopenia.4
AA is associated with CD8+ autoreactive cytotoxic T cells which inhibit haematopoietic stem cells, leading to apoptosis and BMF; a significant reduction in CD4+ regulatory T cells, which are also non-functional and have an abnormal cytokine profile; and an expansion of CD4+ T helper (Th) 1 and Th17 cells.5-7 These anomalies can be identified by routine flow cytometry or cytokine profiling. Recent studies using newer techniques such as single cell mass cytometry (also called Cy-TOF) have identified aberrant subsets of T regulatory cells with pro-inflammatory properties in AA that predict poor response to IST.8
PNH in the context of BMF is characterised by progressive expansion of one or more PNH stem cell clones (immunological escape clones), which are deficient (partial or complete) in the expression of glycosylphosphatidylinositol (GPI)-anchored proteins.9 Other examples of such escape clones include trisomy 8, which leads to expansion of Wilms’ tumour 1-specific cytotoxic CD8+ T cells,10 uniparental disomy on chromosome 6p, the human leukocyte antigen (HLA) locus, and del 13q, which has been increasingly associated with BMF with PNH clones and responsive to IST, particularly in the context of AA rather than MDS.11
Several somatic mutations have been identified in a subgroup of patients with AA that can predict malignant transformation to MDS and monosomy 7.12 In a study of 150 patients with AA and no morphological evidence of MDS, acquired somatic mutations were identified in approximately 20%.12 ASXL1 (8% of patients) and DNMT3A (5% of patients) were the most common mutations, while BCOR was present in 4% of patients. In this study, 41% of patients had <10% mutation clonal load.12 In patients with AA with a disease duration of >6 months, these somatic mutations were associated with a 40% risk of transformation to MDS versus 4% risk without the mutations.12 Similar findings have been reported by the National Institutes of Health (NIH), Cleveland, and Japanese groups.13 The acquisition of these mutations increases with age, with each mutation demonstrating a different pattern of change over time.13 Importantly, the identification of these mutations may allow prediction of response to IST.13 Interestingly, presence of the BCOR mutation seems to predict a better response to IST.12 Additionally, studies investigating the mutational landscape of PNH indicate a stepwise pattern of MDS-type mutations, with these mutations arising either as a sub-clone within the PIG-A mutant population or prior to PIG-A mutation.14
Telomere length/attrition is another important factor in transformation to MDS in AA.12,15 Patients with AA and somatic mutations have been shown to have reduced telomere length. In a study of 13 patients with severe AA, patients who evolved to MDS and AML had marked telomere attrition that preceded the emergence of monosomy 7.15 In a study of patients with refractory AA treated with eltrombopag (a small molecule agonist of the c-mpl [thrombopoietin] receptor), approximately 40% of patients showed an improved haematologic response; however, they also had an increased risk of clonal evolution to monosomy 7, with a shorter time to evolution.16
There are several different ways in which monosomy 7 can occur, including familial origin, myeloid neoplasia, following haematopoietic stem cell transplantation (HSCT), AA, and following treatment with granulocyte colony-stimulating factor.17 In a recently reported Phase I/II study, patients with telomere diseases were treated with the synthetic steroid danazol 800 mg/day for 24 months, with the aim of attenuating accelerated telomere attrition. After just 3 months of treatment, almost all patients had a gain in telomere length and 79% had haematological response. Adverse effects included elevated liver enzymes (41%) and cramps (33%).18
Reports of spontaneous remission in patients with PNH have been described in the literature.19,20 In the UK PNH Service, where 303 patients have been treated with eculizumab (a humanised monoclonal antibody to complement protein C5), five patients have stopped treatment due to spontaneous remission,21 although the recent case report indicates that there is no recovery of normal haematopoiesis.19
A Phase III trial (A Prospective Randomized Multicenter Study Comparing Horse Antithymocyte Globuline [hATG] + Cyclosporine A [CsA] With or Without Eltrombopag as Front-line Therapy for Severe Aplastic Anemia Patients; RACE) is in progress to evaluate the efficacy and safety of eltrombopag versus placebo combined with IST.
Diagnosis and Management of Patients with Aplastic Anaemia
Professor Gérard Socié
AA is characterised by pancytopenia and persistent unexplained marrow hypoplasia. There is no specific marker and the diagnosis is mainly consequent with the differential diagnosis of hypoplastic MDS.22 Careful blood analysis, bone marrow aspirate, and bone marrow biopsy are all important aspects in correct diagnosis.22 Differential diagnosis versus congenital disorders should always be considered, especially with regard to dyskeratosis congenita.23 Finally, differentiation from MDS should be considered, as outlined earlier in this report.
Severe AA is defined as hypocellularity <30% and at least two criteria from: <0.5×109/L polymorphonuclear neutrophils, <20×109/L platelets, and <20×109/L reticulocytes; very severe AA is defined as <0.2×109/L polymorphonuclear neutrophils.24 All patients with severe or very severe AA require treatment.
For patients <40 years of age with severe AA, HSCT is mandatory, as it has been shown to be effective with low treatment-related mortality. At a median follow-up of 73 months in 61 patients (mean age: 21 years) who underwent HSCT from an HLA-matched sibling donor after irradiation-based conditioning with cyclophosphamide and ATG, 6-year overall survival was 87% (95% confidence interval: 78–97).25 Only one patient developed secondary malignancy. Osteonecrosis (observed in 10 patients) remains a concern in this disease.25
In patients >40 years of age or without a sibling donor, IST with ATG plus cyclosporine is an effective alternative to HSCT and improves blood counts and survival. A Phase III randomised trial comparing hATG and rabbit ATG (rATG) in 60 patients with severe AA showed that hATG was superior to rATG as a first-line treatment for severe AA. At 6-month follow-up, the haematologic response was 68% for patients treated with hATG versus 37% with rATG (p<0.001). Overall survival at Day 800 was 86% in the hATG group versus 68% in the rATG group (p=0.009). Transplant-free survival was 52% for rATG versus 76% for hATG (p=0.002).26 Similar results were demonstrated in a Phase II study conducted in 35 patients with AA treated with rATG and compared with 105 age and disease severity-matched patients from the European Blood and Marrow Transplant (EBMT) registry.27 The ongoing RACE study (mentioned earlier) is also investigating hATG with or without eltrombopag as first-line therapy for severe AA.
In patients refractory to IST, the decision to transplant stem cells from unrelated donors often represents a dilemma for physicians. Evaluations of outcomes in patients <30 years of age without a sibling donor and refractory to IST demonstrate that survival after unrelated HSCT for severe AA has improved significantly over the past 15 years, due to better HLA matching and improved conditioning regimens.28 Similar improvements in survival rates have been shown in other studies in patients with severe AA and unrelated donors receiving fludarabine, cyclophosphamide, and ATG.29
For patients >30 years of age and refractory to IST and with no suitable unrelated donor, there are three options: second-line IST, androgens (20–30% response rate for each), or currently still an investigational drug, eltrombopag. Long-term follow-up of a cohort of 43 patients with severe AA treated with eltrombopag demonstrated durable tri and bi-lineage responses, with a 40% response rate at 3–4 months.16
In conclusion, in the diagnosis of AA, inherited AA and MDS must be excluded. First-line treatment remains IST or bone marrow transplant, according to the availability of a donor. In patients who are refractory to first-line therapy, HSCT from a matched unrelated donor may be used in patients <30 years of age, and in patients >30 years of age, eltrombopag, androgen, or second-line IST.
Diagnosis and Management of Paroxysmal Nocturnal Haemoglobinuria in the Context of Bone Marrow Failure
Doctor Alexander Röth
The classical clinical triad of PNH includes haemolytic anaemia, thrombophilia, and cytopenia. Haemoglobinuria is present in approximately one-third of patients at the time of diagnosis. All patients have some degree of BMF, from isolated thrombocytopenia to AA, which typically precedes PNH. Thromboembolic complications, usually involving the brain, liver, or abdomen, are the leading cause of morbidity and mortality in patients with PNH.30-32 PNH originates from a somatic mutation in the PIG-A gene in a HSC, which is essential for biosynthesis of GPI-anchors for proteins, two of which (CD55 and CD59) are complement regulatory proteins. Selection and expansion of the mutant stem cell are both necessary for clinical development of PNH, which may explain the rarity of this disease.33 The absence of CD55 and CD59 on affected red blood cells leads to the formation of a membrane attack complex, which activates complement-mediated haemolysis and the release of free haemoglobin.34 Large amounts of free haemoglobin causes depletion of nitric oxide. The resulting reduction of nitric oxide levels leads to the activation of haemostasis with thromboembolic complications and smooth muscle dystonias that occur in PNH, which manifest as abdominal pain, dysphagia, pulmonary and systemic hypertension, and erectile dysfunction caused by impaired regulation of smooth muscle contractions.35
Patients are typically diagnosed with PNH in their early 30s36 and 5-year mortality is 35%, despite best supportive care.19 Thrombosis occurs in 18–40% of patients with PNH and increases the risk of death 7-fold compared with patients without thrombosis.19,37 Studies have reported a 21% incidence of a thrombotic event prior to PNH diagnosis.19,38 Silent thromboembolic complications are also possible in PNH and may be severe. Evaluation of the entire blood vessel might be helpful for diagnosis and may be conducted using magnetic resonance angiography with the angiographic system for unlimited rolling fields of view (AngioSURF).39
Markers for risk of thrombosis include D-dimers and tissue factor microparticles, both of which may be elevated in patients with PNH.40-43 Patients with thrombocytopenia also have an elevated risk of thromboembolism.40 Data from the South Korean National PNH Registry identified several haemolysis and clinical symptoms associated with increased risk of thromboembolism.37 Patients with elevated haemolysis (lactate dehydrogenase [LDH] levels ≥1.5-times the upper limit of normal [ULN] at diagnosis) were at significantly higher risk for thromboembolism than patients with LDH <1.5-times ULN (odds ratio: 7.0; p=0.013). The combination of LDH ≥1.5-times ULN with the clinical symptoms of abdominal pain, chest pain, dyspnoea, or haemoglobinuria was associated with a greater increased risk for thromboembolism than elevated haemolysis or clinical symptoms alone.37
Flow cytometry is the gold standard for diagnosis of PNH.44 Detection of GPI-anchor proteins such as CD59 or CD55, or fluorescent aerolysin reagent (FLAER) on haematopoietic cells using monoclonal antibodies forms the basis of a specific PNH diagnostic test.44,45 Identification of FLAER may be particularly useful as it selectively binds to the glycan core of GPI.46 Patients at risk who should be tested for PNH include those with haemolysis (e.g. Coombs negative haemolytic anaemia, haemoglobinuria, or renal dysfunction), BMF (e.g. AA, MDS, or cytopenia), and patients with unexplained thrombosis.44,47
Supportive care for patients with PNH may include blood transfusion, folic acid and vitamin B12 supplementation, oral iron supplementation (if the patient is iron deficient), early treatment of bacterial infections with antibiotics, hydration (in critical haemolysis), and anticoagulants (although these may be non-effective in many patients).44,48-50 Supportive care however, does not affect disease progression and may be associated with additional adverse risk factors.
The monoclonal antibody eculizumab blocks the terminal complement cascade, eliminates the formation of the membrane attack complex, and prevents cell lysis.51,52 In clinical trials, eculizumab improves survival and has demonstrated significant efficacy in reducing haemolysis, the need for blood transfusions, and the risk of thromboembolic complications versus controls. Patients treated with eculizumab also had reduced levels of fatigue, independent of haemoglobin levels, and a lower incidence of renal complications.30,38,53-59 Eculizumab is indicated in adults and children for the treatment of patients with PNH and atypical haemolytic uraemic syndrome (aHUS).29 In patients ≥40 kg, the recommended dose of eculizumab is 900 mg every 14±2 days.30,60 Patients on eculizumab are susceptible to meningococcal infections, and vaccination with Meningococcal Group A, C, W135, Y conjugate vaccine (e.g. Menveo®) followed by Meningococcal Group B Vaccine (Bexsero®) should be considered.58 Predictors of response to eculizumab include the degree of underlying BMF, concurrent inflammatory conditions such as bacterial and viral infections that can cause breakthrough haemolysis and autoimmune disease such as Crohn’s disease, genetic factors such as missense C5 heterozygous mutation, and polymorphisms of CR1 that lead to a suboptimal response due to extravascular haemolysis. Figure 2 shows a proposed treatment algorithm for PNH; in the haemolytic setting, symptomatic patients can be treated with eculizumab in combination with supportive therapy.61
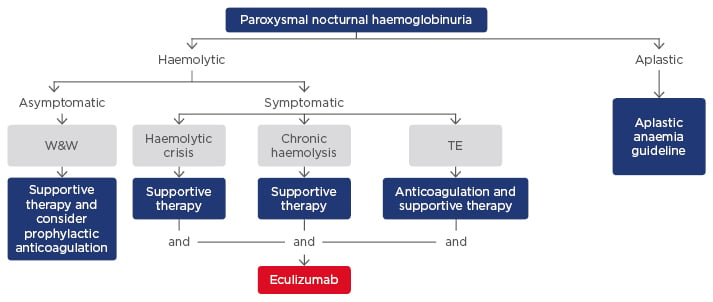
Figure 2: Proposed treatment algorithm for paroxysmal nocturnal haemoglobinuria.61
TE: thromboembolic event; W&W: wait and watch.
In a recently published case-report, a 64-year old female with PNH, thromboembolic complications, and subsequent transition to severe AA was treated with IST (hATG + cyclosporin A) in combination with eculizumab. No reduced ATG efficacy or severe adverse events were observed. Re-occurrence of PNH symptoms was prevented, while T cell depletion was similar to non-eculizumab-treated patients, with partial remission being evident by Day 83.62
In conclusion, PNH is a chronic, life-threatening disease associated with chronic complement-mediated haemolysis. Thrombosis is multifactorial, may occur unexpectedly, and is the primary cause of death in patients with PNH. The gold standard test for PNH is high-sensitivity flow cytometry performed on peripheral blood. Terminal complement inhibition with eculizumab has become the standard of care for symptomatic PNH.





