![EMJ Oncology 9 [Supplement 8] 2021 Feature Image](https://www.emjreviews.com/wp-content/uploads/2021/11/EMJ-Oncology-9-Supplement-8-2021-Feature-Image-940x563.jpg)
Chairperson: Anna Sureda1
Interviewees: Jason Westin,2 Georg Hess3
1. Institut Català d’Oncologia-Hospitalet, Barcelona, Spain
2. University of Texas, MD Anderson Cancer Center, Houston, Texas, USA 3. University Medical School of Johannes, Gutenberg University, Mainz, Germany
Disclosure: Sureda has a financial interest/relationship or affiliation in the form of consultant for Amgen Inc., Bristol Myers Squibb Company, Celgene Corporation, F. Hoffmann-La Roche Ltd., Gilead Sciences, Inc., GlaxoSmithKline plc., Janssen Inc., Kite Pharma, Inc., Novartis AG, Sanofi, and Takeda Pharmaceutical Company Limited; speakers bureau participant with Takeda Pharmaceutical Company Limited; honoraria from Amgen Inc., Bristol Myers Squibb Company, Celgene Corporation, F. Hoffmann-La Roche Ltd., Gilead Sciences, Inc., GlaxoSmithKline plc., Kite Pharma, Inc., Novartis AG, Sanofi, and Takeda Pharmaceutical Company Limited; and advisory board for Amgen Inc., Bristol Myers Squibb Company, Celgene Corporation, F. Hoffmann-La Roche Ltd., Gilead Sciences, Inc., GlaxoSmithKline plc., Janssen Inc., Kite Pharma, Inc., Novartis AG, Sanofi, and Takeda Pharmaceutical Company Limited. Westin has a financial interest/relationship or affiliation in the form of consultant for ADC Therapeutics SA, AstraZeneca, Bristol Myers Squibb Company, Forty Seven, Inc., Genentech, Inc., Iksuda Therapeutics Ltd., Janssen Inc., Kite Pharma, Inc., MorphoSys AG, and Novartis AG; and grant/research support from ADC Therapeutics SA, AstraZeneca, Bristol Myers Squibb Company, Forty Seven, Inc., Genentech, Inc., Janssen Inc., Kite Pharma, Inc., MorphoSys AG, and Novartis AG. Hess has a financial interest/relationship or affiliation in the form of: consultant for AstraZeneca, F. Hoffmann-La Roche Ltd, Genmab A/S, Gilead Sciences, Inc., Incyte, and Janssen Inc.; grant/research support from Celgene Corporation, F. Hoffmann-La Roche Ltd, Janssen Inc., and MorphoSys AG; honoraria from AbbVie Inc., AstraZeneca, Bristol Myers Squibb Company, F. Hoffmann-La Roche Ltd., Genmab A/S, Gilead Sciences, Inc., Incyte, and Janssen Inc; and advisory board for AstraZeneca, F. Hoffmann-La Roche Ltd., Genmab A/S, Gilead Sciences, Inc., Incyte, and Janssen Inc.
Acknowledgements: Medical writing assistance was provided by Katherine Kahn, Holyoke, Massachusetts, USA, and PeerVoice.
Support: This symposium was funded by an unbiased educational grant from Incyte Biosciences International Sàrl. The views and opinions are those of the authors and not necessarily those of Incyte Biosciences International Sàrl or PeerVoice.
Citation: EMJ Hematol. 2021;9[Suppl 6]:2-12.
Interview Summary
Although approximately 60% of patients with diffuse large B-cell lymphoma (DLBCL) can be cured, the remaining 40% have relapsed or refractory disease (R/R DLBCL). Substantial unmet need remains for effective, safe therapies for these patients. Novel targeted approaches to the treatment of R/R DLBCL include CD19-directed antibodies, antibody–drug conjugates, bispecific monoclonal antibodies, and anti-CD-19 chimeric antigen receptor (CAR) T-cell therapies. Recent clinical trial data have demonstrated robust responses for these newer agents, with acceptable safety profiles. However, patient selection must be carefully considered when choosing therapies in R/R DLBCL, based on disease characteristics, patient factors and preferences, and whether a curative approach is feasible. Treatment sequencing algorithms now allow for easier patient stratification to identify whether intensive therapy or a non-curative approach is the best option.![]()
Erratum: Immunotherapies for Relapsed/Refractory Diffuse Large B-Cell Lymphoma: Which Antibodies for Which Patients?
Speakers:Anna Sureda, Jason Westin, Georg Hess
Original citation:EMJ Oncol. 2021;9[Suppl 8]:2-12.
Date correction published: 30.11.21
The article that included Sureda et al. in Suppl 8 of EMJ Oncology 9.1 (pages 2-12) was originally published on 12.11.2021. Since then an erratum has been made. The citation “EMJ Hematol. 2021;9[Suppl 6]:2-12” originally read “EMJ Oncol. 2021;9[Suppl 8]2-12” incorrectly. It has now been updated.
The EMJ apologises for the error and any inconvenience caused.
![]()
INTRODUCTION
DLBCL is the most common histological subtype of B-cell neoplasia and accounts for 37% of patients with newly diagnosed non-Hodgkin lymphoma.1 DLBCL is genetically and phenotypically a heterogeneous entity, with substantial variation in long-term outcomes among different patient subgroups.2,3 With the standard treatment approach of rituximab in combination with cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP), approximately 60% of patients with DLBCL can be cured.4 However, 20–30% will relapse after first complete remission and 10–15% will have primary refractory disease.5 Of the approximately 50% of patients eligible for autologous stem cell transplantation (SCT), only 40% will be cured by transplant.5 Therefore, substantial unmet need remains for effective, safe therapies for patients who are transplant-ineligible, relapse after transplantation, or have an inadequate response to first-line salvage chemotherapy.
In recent years, efforts to improve long-term outcomes in these patients have led to strategies that alter R-CHOP, such as adding a new targeted agent (e.g., bortezomib, ibrutinib, or lenalidomide), replacing a component (e.g., with bortezomib or obinutuzumab), or consolidating remission with maintenance therapy (e.g., rituximab, lenalidomide, enzastaurin, or everolimus).6–14 Unfortunately these efforts have been largely unsuccessful in improving survival in R/R DLBCL.
European Society for Medical Oncology (ESMO) and National Comprehensive Cancer Network (NCCN) guidelines recommend the use of platinum- or gemcitabine-based chemotherapeutic protocols for first relapse or progression of DLBCL in both transplant-eligible and transplant-ineligible patients.15 Consolidation is recommended with autologous SCT; allogenic SCT can be considered for patients that relapse after autologous SCT or that have poor risk features at time of first relapse. Enrolment in a clinical trial should be considered as an option in R/R DLBCL.15,16
Novel Immune Targets in Refractory or Relapsing Diffuse Large B-Cell Lymphoma
A variety of promising therapeutic agents in R/R DLBCL target cell surface antigens, including CD19, CD20, CD22, CD3, and CD79b.17 CD19 is a transmembrane protein uniformly expressed over the maturation process of the B-cell lineage, making it a promising target for several new treatment strategies in patients with R/R DLBCL.18 Novel targeted approaches in R/R DLBCL include naked antibodies (e.g., rituximab, obinutuzumab, tafasitamab) targeting cell surface antigens, antibody-drug conjugates (e.g., polatuzumab vedotin) that are capable of delivering a high quantity of cytotoxic agent into malignant B cells, and bispecific monoclonal antibodies (e.g., blinatumomab) that enhance immune cells to attack tumour cells, and anti-CD19 chimeric antigen receptor (CAR) T-cell therapies (Figure 1).
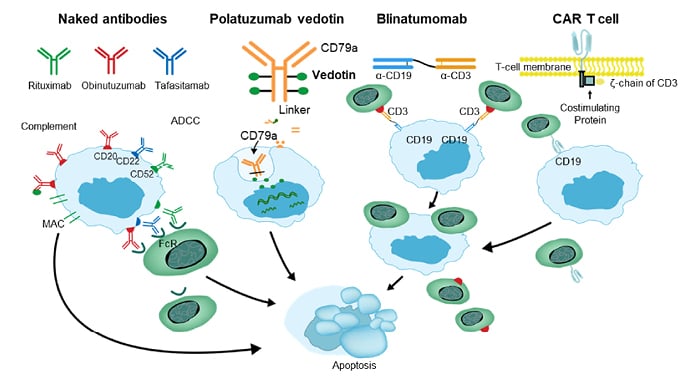
Figure 1: Immune targets of interest for patients with relapsing or refractory diffuse large B-cell lymphoma
ADCC: antibody-dependent cellular cytotoxicity; CAR: chimeric antigen receptor; FcR: Fc receptor.
Immune escape mechanisms frequently evolve in malignant B cells of DLBCL.19 Immune checkpoint inhibitors, radioimmunotherapy, antibody–drug conjugates, and CAR T-cell constructs are able to circumvent the escape mechanisms of malignant B cells in DLBCL. Novel agents are in development that also target well-known molecular signalling pathways, such as VEGFR, BCR pathways, JAK/STAT 3 pathways, and epigenetic regulars, such as HDAC, BET, and EZH2.17 These novel agents are being evaluated in numerous Phase I and Phase II prospective clinical trials, with some notable results in terms of overall response and complete remission rates (Table 1). This review, based on a symposium held by PeerVoice, will cover efficacy and safety from the latest clinical trial data on promising treatment approaches and their potential roles in second- and third-line treatment strategies for R/R DLCBCL, with a special focus on anti-CD19 therapies.
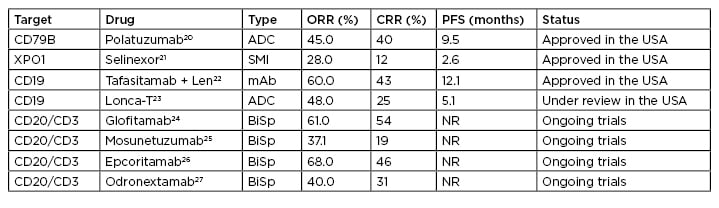
Table 1: Targeted therapy on the horizon for diffuse large B-cell lymphoma.
ADC: antibody-drug conjugates; BiSp: bispecific; CRR: complete response rate; mAb: monoclonal antibodies; NR: not reported/reached; ORR: overall response rate; PFS: progression-free survival; SMI: small molecule inhibitor.
EMERGING STRATEGIES IN RELAPSED OR REFRACTORY DIFFUSE LARGE B-CELL LYMPHOMA
CD19-Targeted Naked Antibodies: Tafasitamab
The Fc-enhanced, humanised anti-CD19 monoclonal antibody tafasitamab results in direct cytotoxicity via binding at the CD19 site of malignant B cells, as well as antibody-dependent cellular cytotoxicity and phagocytosis.28 Tafasitamab has received European Medicines Agency (EMA) and U.S. Food and Drug Administration (FDA) approval in combination with lenalidomide for the treatment of primary R/R DLBCL in transplant-ineligible patients.28
The L-MIND trial is an ongoing, Phase II single arm, open-label multicentre study of tafasitamab in combination with lenalidomide in patients with R/R DLBCL who are transplant-ineligible. Data from the L-MIND trial was recently presented at the American Society of Hematology (ASH) 2020 annual meeting.29 In the first three treatment cycles, tafasitamab was dosed once weekly, then every 2 weeks during Cycles 4–12. After Cycle 12, patients who were progression-free received tafasitamab every 2 weeks until disease progression. Lenalidomide was administered on Days 1–21 at a dose of 25 mg.29 The primary endpoint was the objective response rate (ORR). Secondary endpoints included duration of response (DoR), progression free survival (PFS), overall survival (OS), and safety. In the 80 enrolled patients, ORR was 58.8%; 41.3% had a complete response (CR), and 17.5% had a partial response (PR).29 An evaluation of long-term outcomes found the median DoR in patients with CR or PR was 34.6 months, representing a sustained response for patients with DLBCL (95% confidence interval [CI]: 26.1–34.6).30 Median PFS was 16.2 months (95% CI: 6.3–not reported) and OS was a median of 31.6 months (95% CI: 18.3–not reported). These results are promising, given expected outcomes with chemotherapeutic options in patients with R/R DLBCL.29,30
Also, these positive responses occurred in the setting of substantially less toxicity than that observed with conventional chemotherapy regimens. The most frequently reported grade ≥3 haematologic treatment-emergent adverse effects (TEAEs) were neutropenia in 49.4%, thrombocytopenia in 17.3%, and febrile neutropenia in 12.3% of patients.30 However, haematologic TEAEs were far less likely when patients transitioned to tafasitamab monotherapy after completing the combination treatment phase.30
The global, observational cohort study, Re-MIND, endeavoured to estimate the tafasitamab contribution to the outcomes achieved with tafasitamab/lenalidomide.31 The trial collected data from multiple international centres of 490 patients treated with lenalidomide monotherapy for R/R DLBCL.31 Of the 490 patients, baseline covariates of 76 patients were matched with 76 patients from the original L-MIND dataset. The primary endpoint was difference in ORR, with key secondary endpoints including differences in CR rate, PFS, DoR, and OS.31 The best ORR in the tafasitamab/lenalidomide group was 67.1%, with a CR rate of 39%, whereas the best ORR and CR rate in the lenalidomide monotherapy group were 34.2% and 13%, respectively (95% CI: 23.7–46.0). The ORR odds ratio was 3.9 (95% CI: 1.9–8.1; p<0.0001), favouring the tafasitamab/lenalidomide combination group.31 PFS was also significantly longer among patients treated with combination therapy, with a median PFS of 12.1 months, compared to 4 months in the monotherapy group. Furthermore, OS also favoured the L-MIND cohort, with a hazard ratio (HR) of 0.499 (95% CI: 0.317–0.785; p=0.0026).31 Other studies have shown that lenalidomide monotherapy or in combination with rituximab have an ORR response rate of approximately 35%,32,33 suggesting that the combination of tafasitamab/lenalidomide appears to confer greater efficacy than lenalidomide with any random antibody.
Antibody–Drug Conjugates: Loncastuximab tesirine
Loncastuximab tesirine is a novel antibody–drug conjugate composed of a humanised CD19-targeted monoclonal antibody and conjugated via a linker to a pyrrolobenzodiazepine-dimer toxin.17 Once bound to a CD19-expressing cell, loncastuximab tesirine is internalised into the cell where enzymes cleave the linker, releasing the pyrrolobenzodiazepine dimers. Cytotoxic DNA cross-links are formed, stalling the DNA replication fork and resulting in cell apoptosis.17 A recently completed Phase I first-in-human trial demonstrated encouraging antitumour activity and an acceptable safety profile in R/R DLBCL.34 At the 25th Annual Meeting of the European Hematology Association (EHA) in 2020, initial results were presented of a Phase II, single-arm, open-label trial evaluating the efficacy of loncastuximab tesirine as a single agent in 145 patients with R/R DLBCL.35,36 Enrolled patients received 150 µg/kg loncastuximab tesirine every 3 weeks for the first two cycles, followed by 75 µg/kg every 3 weeks for subsequent cycles up to 1 year. If patients still derived benefit from loncastuximab tesirine at 1 year, they could subsequently receive it every 12 weeks to derive additional benefit beyond 1 year.35,36 Patients had an ORR of 48.3% (95% CI: 39.9–56.7), with a CR rate of 24.1%. An additional 15.2% had stable disease. Moreover, in a small subset of patients (N=11) with high-grade B-cell lymphomas (also known as ‘double-hit’ lymphomas), the ORR was 45.5%.35 Although data on the DoR is still accumulating, the median DoR as of April 2020 was 1025 months, with no precipitous decline, suggesting that a substantial proportion of patients have a durable response to treatment.35
Given that loncastuximab tesirine is essentially a delivery device for a chemotherapeutic agent, it is not surprising to find that TEAEs were similar to chemotherapy and included neutropenia, thrombocytopenia, fatigue, and nausea.35 It is not yet known if these side effects are potentially cumulative, which could limit the utility of loncastuximab tesirine as a long-term therapy. Uniquely, gamma glutamyl transferase was elevated in approximately 41% of patients, whereas alkaline phosphatase was elevated in just 20% of patients, suggesting that gamma glutamyl transferase elevation is a specific TEAE of loncastuximab tesirine treatment.35
Antibody–Drug Conjugates: Polatuzumab Vedotin
Polatuzumab vedotin is an antibody–drug conjugate targeting CD79b, a cell surface marker that is expressed in >95% of patients with DLBCL.37,38 Polatuzumab vedotin delivers the microtubule inhibitor monomethyl auristatin E and has demonstrated activity in R/R DLBCL as monotherapy.20,39 It has also been evaluated in combination with an anti-CD20 monoclonal antibody, with ORRs ranging from 13–56%.40 However, CR rates are no higher than 15%, leading to investigations combining polatuzumab vedotin with additional agents to improve these outcomes. A recently published randomised, Phase II trial examined the efficacy of polatuzumab vedotin combined with bendamustine and rituximab (BR) versus BR in patients with R/R DLBCL.20 Patients in the polatuzumab/BR arm had significantly longer median OS (12.4 months versus 4.7 months, HR: 0.42; 95% CI: 0.24–0.75; p=0.002) and median PFS (9.9 months versus 3.7 months; HR: 0.36; 95% CI: 0.21–0.63; p<0.001).20 Despite these impressive outcomes, it is uncertain how much benefit the bendamustine adds to the polatuzumab/rituximab combination. In general, bendamustine is not commonly used in patients with R/R DLBCL. An earlier study from 2013, in which BR was used as doublet therapy in 59 patients with R/R DLBCL, reported a relatively high CR rate of 37% and a PR rate of 25%. However, most patients in this study had a less aggressive, late-relapsing disease than is typically seen in clinical trials.41 In the nine patients who relapsed within 12 months of their prior treatment, response rates were less impressive, with a CR rate of 0% and 44% achieving a PR.41
Antibody immunomodulatory agents and antibody-drug conjugates confer some practical benefits that may be important in individualising therapeutic choices in R/R DLBCL, including immediate availability, rather than the need to wait for manufacturing, a process that may potentially be too long for a patient with aggressive refractory disease. Moreover, these therapies are logistically simple. They can be administered in any oncology centre, with no special expertise required for managing cell therapy toxicities or need of monitoring programmes for cytokine release syndrome or neurologic toxicities, a potential drawback of CAR T-cell therapy. Additionally, there is now decades of experience combining antibodies with both chemotherapy and targeted therapies; while cell therapies in combination with other agents may have advantages in treating malignancies, the additional agents, particularly chemotherapeutic agents, may potentially have a negative effect on the health of CAR T-cells.
SELECTING PATIENTS FOR IMMUNOTHERAPY
If untreated, R/R DLBLC is a rapidly fatal disease. As discussed earlier, approximately 60% of patients with DLBCL achieve a complete cure with R-CHOP; however, approximately 20–30% of treated patients relapse and approximately 10–15% have refractory disease.42 Most patients who relapse will do so within 2 years after initial therapy, but 7% of patients relapse more than 5 years after treatment.43 Patients experiencing no relapses have been shown to have a near-normal life expectancy, whereas survival probability falls dramatically for those with early relapsing DLBCL.44 The minority of patients with late relapse have somewhat better outcomes.44
Drawing from two large randomised trials and two academic databases, the SCHOLAR-1 study was the first patient-level analysis of outcomes in refractory DLBCL and identified poor outcomes in this patient population.45 SCHOLAR-1 found an ORR of 26% with a CR of 7% to the next line of therapy, with a median OS of 6.3 months.45 Only 20% of patients with refractory DLBCL were alive at 2 years; most of these patients had received autologous SCT with a CR or PR; however, the majority of patients (73%) were not able to receive SCT or did not respond to salvage therapy.45 Consistently poor outcomes were observed across all patient subgroups and study cohorts.45
Potential options for curative therapy in R/R DLBCL include high-dose therapy, allogenic SCT, and CAR T-cell therapy. The landmark PARMA trial established high-dose therapy as superior to conventional treatment, but success in patients relapsing within the first year is low.46 CAR T-cell therapy uses gene transfer technology to reprogramme a patient’s T-cells to express CARs, directing the cytotoxic potential of T-cells against tumour cells.47 CARs are developed from fusion proteins containing an extracellular antigen-binding domain. The domain consists of a single-chain variable fragment derived from an antibody and intracellular signalling domain involved in initiating T-cell signalling and T-cell effector functions.47 Newer second-generation CARs contain costimulatory domains that dramatically increase CAR T-cell persistence and antitumor efficacy.47 Using leukapheresis, antibody-coated beads are added to the patient’s lymphocytes, then the modified T-cells are expanded. Following bead removal, these cells are then reinfused into the patient.47
Clinical trials have demonstrated that CAR T-cell treatment induces long-term remissions in approximately 40–50% of patients.48–50 Axicaptagene ciloleucel was evaluated in the ZUMA Phase I/II trial. Of 101 patients, 83% treated with axicabtagene ciloleucel for refractory DLBCL achieved an objective response and 58% of patients achieved a CR, with a median DoR of 11.1 months.48 Tisagenlecleucel was evaluated in the JULIET trial. Of 115 infused patients, the ORR was 54% (95% CI: 43–64%), with a 40% CR rate and 13% PR rate.49 The ORR was consistent across prognostic subgroups, including those with prior autologous SCT and double/triple-hit lymphoma.49 The probability of being relapse-free was 66% (95% CI: 51–78%) at 6 months and 64% (95% CI: 48–76%) at 12 and 18 months. OS probability was 48% (95% CI: 38–57%) at 12 months and 43% (95% CI: 33–53%) at 18 months.49 In the TRANSCEND trial, lisocabtagene maraleucel was evaluated in heavily pretreated patients with aggressive disease.50 The ORR was 73% (95% CI: 67–78%), with a CR rate of 53% (95% CI: 47–59%); responses were similar across patient subgroups.50 Median DoR, PFS, and OS were 13.3 months, 6.8 months, and 19.9 months, respectively.50 Despite these positive outcomes, studies have shown that patients with multiple lines of treatment, bulky disease, and high serum lactate dehydrogenase are less likely to have a beneficial treatment response to CAR T-cell therapies.51,52
In predicting curability in patients with R/R DLBCL, individual patient-, disease-, and treatment-related factors must be considered. Only 40% of those who relapse or progress while on frontline treatment are eligible for autologous SCT or CD19-directed CAR T-cell therapy, based on their ability to withstand aggressive treatment. It is also crucial to assess whether disease allows for curative treatment (i.e., there is no fulminant disease course or other factors pointing to rapid disease progression). Treatment history must also be considered, including response or lack of response to prior treatment attempts, as well as earlier conflicting treatments that may impair T-cell fitness (e.g., bendamustine) if CAR T-cell therapy is an option.
In selecting appropriate curative options, both age and fitness must be considered. The Eastern Cooperative Oncology Group (ECOG) Performance Status incorporates both parameters and can identify patients eligible for autologous or allogenic SCT or palliative care.16 In general, transplantation is less likely to be appropriate in patients older than 65–70 years. CAR T-cell therapy, however, is now recognised to be a reasonable option in patients over the age of 70 if the ECOG score is ≤1. In addition to ECOG score, other methods that define patient risk must be considered. The Hematopoietic Cell Transplantation-Comorbidity Index (HCT-CI) provides validated and reliable scoring of pretransplant comorbidities associated with non-relapse mortality and survival to assess risk before allogenic HCT.53–55 Definition of high-risk clinical features, such as serum LDH, is also a key step in choosing treatments. The International Prognostic Index (IPI) and age adjusted IPI are widely used tools that were recently shown to help guide selection of patients who would benefit from CAR T-cell therapy and to predict toxicities or outcomes.56
In patients with R/R DLBCL who are eligible for a curative approach, CD19-targeting drugs should be avoided prior to CAR T-cell treatment, due to their potential negative effects on T-cell health. Also, bendamustine should not be used at relapse because of its negative effects on T-cell function. Also, the role of antibody–drug conjugates as a bridging treatment is currently poorly understood, especially since they are frequently used with BR. Moreover, there is only limited understanding of the role of bispecific antibodies as bridging treatment and effects on CAR T-cell therapies.
Unfortunately, just one-half of the patients with relapsed DLBCL have curative options, even after high-dose therapy, stem cell transplant, or CAR T-cell therapy, more than 80–85% will not be cured by these strategies.3 In patients who cannot undergo curative treatments, achievable goals of therapy include long-term disease control, sustained quality of life, and, in a small subset of patients, CAR T-cell therapy.15,16
New Agents and Regimens for Transplant-Ineligible Patients
Treatment strategies for transplant-ineligible, non-curative patients have evolved over the last decade. As discussed earlier in this review, tafasitamab combined with lenalidomide and antibody–drug combinations have shown the potential for high response rates with superior survival benefits.20,29,30,35 Additionally, numerous bispecific antibodies are now available, including mosunetuzumab, epcoritamab, glofitamab, and odronextamab. As an example, epcoritamab has demonstrated an ORR of 66.7–100.0% of patients with R/R DLBCL, depending on dose percent, with a CR rate of approximately 30%, a substantial improvement over conventional treatments.57 The small molecule nuclear export inhibitor selinexor, available in some countries, has demonstrated an ORR of 29% and CR of 12% in highly refractory patients.58 The chemotherapy option of pixantrone has an ORR of 37%, a CR of 11%, but a short PFS.59 The traditional approach of gemcitabine/oxaliplatin has a high ORR but a short event-free survival and is not well-tolerated by many patients.60
Considering available treatments, how can clinicians integrate this information into individualised decision-making? Route, frequency, and duration of medication administration are important factors that can affect patient quality of life. Tafasitamab is given intravenously, first weekly and then biweekly for maintenance. Polatuzumab vedotin in combination with BR is also administered intravenously for up to six cycles. Bispecific antibodies are administered until disease progression, typically every 3 weeks. Selinexor has a key advantage of being administered orally. Some approaches require inpatient treatment and have higher risk for cytokine release syndrome and neurotoxicity.
Disease characteristics must also be taken into consideration. Currently, molecular genetics cannot reliably determine therapeutic response, but relapse patterns and the presence of comorbidities offer important information and guide treatment selection:
- In central nervous system relapse, a methotrexate-based regimen is warranted.
- If the patient has neuropathy, platinum- and antibody-drug conjugates should be avoided.
- In renal failure, platinum-based therapies are contraindicated and lenalidomide should be avoided or used with dosage adjustments.
- Chemotherapy is generally contraindicated in patients with hepatic failure.
- If the patient has had thromboembolism, anticoagulation should be considered if lenalidomide is used.
- With active neurologic impairment, bispecific antibodies must be used with caution.
Individual patient preferences must also be considered, including patient goals and the capacity to accept and tolerate adverse effects of treatment. Functional scores, such as ECOG and the Charlson Comorbidity Index (CCI), can be incorporated to guide treatment decision-making.
Prior treatment also influences choice of therapy. A patient whose disease is entirely refractory to chemotherapy or has a short duration of remission is likely to have poor response to subsequent chemotherapeutic agents; therefore, other types of treatment should be considered. Extensive use of CD20 monoclonal antibodies requires assessment to determine continuing CD20 positivity, especially prior to the use of bispecific antibodies targeting CD20. As mentioned previously, prior use of bendamustine affects T-cell fitness; if CAR T-cell therapy might be considered as an option, clinicians should consider collecting lymphocytes prior to its use or consider other options. Treatment approaches after failure of CAR T-cell therapy are limited; however, one strategy is to use tafasitamab and lenalidomide. However, CD19 positivity should be assessed beforehand to ensure the likelihood of treatment response.
Individualised Treatment Algorithms
Treatment sequencing algorithms have been refined so that patients can be more easily stratified into those for whom intensive treatment is or is not an option (Figure 2). For those in whom intensive treatment is not feasible, there are now viable therapies including bispecific antibodies, single-agent antibodies and immunoconjugates, and small molecules. Antibody-based therapy has paved the way for reasonable treatment options for these patients, allowing for sustainable disease control and a more positive course when a curative approach is not an option.
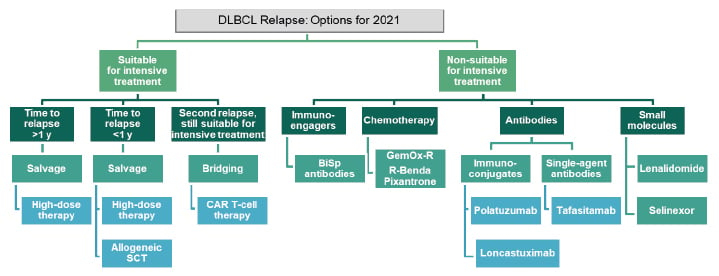
Figure 2: Optimal treatment sequencing for relapsing or refractory diffuse large B-cell lymphoma.
DLBCL: diffuse large B-cell lymphoma; GemOx-R: rituximab, gemcitabine, oxaliplatin; R-Benda: rituximab-bendamustine; SCT: stem cell transplantation.
CONCLUSION
The therapeutic landscape of R/R DLBCL has evolved substantially, offering new approaches for patients whose only treatment options have, until recently, been limited to supportive care. Clinical trial data on CD19-targeted naked antibodies, antibody-drug combinations, and CAR T-cell therapy suggest strong, durable responses in patients with R/R DLBCL. Moreover, a variety of targeted immune therapies can now be used in patients for whom a curative, intensive approach is not an option, offering the potential for sustained disease control.





