
Meeting Summary
The Kite, a Gilead Company, symposium “CAR-T: From bed to bench and back again” took place during the 2019 meeting of the International Conference on Malignant Lymphoma (ICML) in June. It was introduced by Chairperson Dr Roddie, who defined the meeting objectives: to provide an understanding of the science and key translational findings behind chimeric antigenic receptor (CAR) T-Cell development and to discuss the manufacturing process for the CAR T-cell product and the challenges involved in scaling this process from the laboratory to a commercial setting. Mr Rossi presented on the autologous anti-CD19 CAR T-cell therapy development journey and highlighted how collaborations drove this, with academia and the National Cancer Institute (NCI) being under a co-operative research and development agreement (CRADA), and discussed the pivotal ZUMA-1 clinical trial. Dr Roddie followed by describing CAR T-cell therapy development from patient selection to apheresis, highlighting the importance of patient selection and the need for consensus guidance to standardise apheresis between centres. Dr Shen and Mr van de Wiel discussed the manufacturing process for axicabtagene ciloleucel▼ production and how its patient-focussed nature has led to a drive to reduce product turnaround times, leading to manufacturing network expansion in Europe. This talk was followed by a presentation by Prof Chabannon, who explored how product release testing is vital to ensure drug product quality, highlighting how the CAR T-cell manufacturing process differs from that of conventional drug products. He also noted the importance of release tests and quality control (QC) and discussed out-of-specification (OOS) products, and how these can be managed. Finally, Dr Roddie discussed pretreatment patient management and the infusion process, noting that lymphodepleting chemotherapy is critical to the CAR T-cell administration algorithm. She concluded by discussing the challenges in ‘scaling out’ CAR T-cell manufacture to meet patient needs, particularly good manufacturing practice (GMP) cell manufacturing and scaling out quality systems to ensure a standardised process for high-quality treatment in all qualified sites.
Cell Therapy Development Journey: From Bench to Bedside
Mister John Rossi
The initial CAR concept was developed as early as the 1980s.1 Later, CAR research took place under an accelerated timeline. In 2010, the NCI published the first case report of successful anti-CD19 CAR T-cell therapy in a patient with non-Hodgkin’s lymphoma, demonstrating for the first time antitumour efficacy and also an on-target off-tumour effect of prolonged B-cell depletion after therapy.2 Following this initial study, Kite and NCI entered into a CRADA relating to an initial study in a cohort of 22 patients who were treated with an anti-CD19 CAR, preceded by low-dose chemotherapy.3-5 The overall response rate was 73%, with a complete response seen in 55% of patients. In an extended follow-up of the NCI trial, four of five patients with initial complete remission had long-term duration of remission ranging from 38 to 56 months.6 The U.S. Food and Drug Administration (FDA) granted the ‘breakthrough’ biologic license application of axicabtagene ciloleucel on 18th October 2017.
The pivotal ZUMA-1 study started in April 2015. This Phase II multicentre study included 101 patients in two cohorts: the first with refractory diffuse large B-cell lymphoma (DLBCL [cohort 1; n=77]) and the second with refractory primary mediastinal large B-cell lymphoma (PMBCL) or transformed follicular lymphoma (cohort 2; n=24). Key eligibility criteria were no response to last chemotherapy or failed autologous stem cell transplant (relapse within 12 months post-autologous stem cell transplant), and prior therapy with anti-CD20 monoclonal antibody and anthracycline.7 Cells were successfully manufactured for 99% of the patients enrolled and 91% of those enrolled were dosed, demonstrating product reliability and therapeutic feasibility.8,9 After 1 year, 82% of the patients who received infusions showed an objective response and 54% of the patients showed a complete response. Grade 3 or higher cytokine release syndrome (CRS) and neurologic events occurred in 13% and 28% of the patients, respectively.9 A subsequent analysis showed durable responses with a median follow-up of 27.1 months, with 39% of patients still having an ongoing response. Median time to response was 1 month (interquartile range: 1–1). The median overall survival (OS) was not reached with a median follow-up of 27.1 months (95% confidence interval [CI]: 12.8, not estimable [NE]), and the OS rate was 51%; the stability of the Kaplan–Meier curve for OS probability at the 0.5 point demonstrates the effectiveness of CAR T-cell technology in this very ill patient population (Figure 1). At the 2-year data analysis, Grade 3 or higher CRS or neurological events were reported in 11% and 32% of patients, respectively. No new axicabtagene ciloleucel-related CRS or neurological events were reported in the extended analysis at 2 years. Grade 3 or higher infections occurred in 28% of patients. Grade 3 or higher neutropenia, thrombocytopenia, and anaemia adverse events occurred in 39%, 24%, and 46% of patients, respectively. The rates of the identified risks (CRS, neurologic events, infections, and cytopenias) were similar to those in the primary analysis.8,10
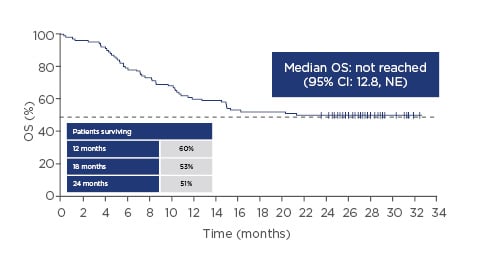
Figure 1: Overall survival over time with axicabtagene ciloleucel.
CI: confidence interval; NE: not estimable; OS: overall survival.
Adapted from Locke FL et al.8
The results showed that early expansion of anti-CD19 CAR T cells (measured by anti-CD19 CAR T-cell peak blood levels within the first 7–14 days) positively correlate with objective responses and neurological adverse reactions.9,10 Rapid expansion of CAR T cells from baseline was seen within 7–14 days after infusion. By 24 months, 11 (34%) of the 32 assessable patients maintained ongoing responses but no longer had detectable gene-marked CAR T cells.
On the other hand, 75% of the assessable patients with ongoing responses showed evidence of B-cell recovery, and initiation of B-cell recovery was noted in some patients as early as 9 months after infusion,8 suggesting that a patient would not need to have functional CAR T cells present to maintain a durable remission. In the USA, axicabtagene ciloleucel received approval from the FDA in October 2017.11 Axicabtagene ciloleucel, marketed as Yescarta,® is a diverse polyfunctional product that is clinically effective across a broad range of CD4:CD8 ratios and comprises phenotypically naïve, central memory, effector, and effector memory T cells.5,9 Yescarta showed clinical efficacy across subsets with poor prognosis, including age, disease state (III/IV), high International Prognostic Index (IPI), bulky disease, cell-of-origin, and double-/triple-hit lymphoma.8,9 European Union (EU) marketing authorisation was received in August 2018 for the treatment of adult patients with relapsed or refractory DLBCL and PMBCL after two or more lines of systemic therapy.10
In summary, ZUMA-1 has a median follow-up in lymphoma patients of 27.1 months8,9 and the OS was not reached. Durable responses were observed at the 2-year follow-up analysis. Two-year analysis of the multicentre ZUMA-1 trial shows that a substantial proportion of patients with refractory LBCL treated with axicabtagene ciloleucel can achieve durable responses with manageable long-term safety.8
From Patient to Commercial Production: Apheresis to Sample Shipping
Doctor Claire Roddie
Patient selection is a vital part of the CAR T-cell production process.12 Patients eligible for treatment with axicabtagene ciloleucel are those with relapsed/refractory DLBCL or PMBCL post two or more lines of systemic therapy. In the pivotal ZUMA-1 trial, patients were required to have adequate organ function, creatinine clearance (Cockcroft–Gault) ≥60 mL/min, serum alanine/aspartate transaminase ≤2.5 upper limit of normal, total bilirubin ≤1.5 mg/dL, ejection fraction ≥50%, no clinically significant ECG findings, no clinically significant pleural effusion, and baseline oxygen saturation >92% in room air.9 Further, it has been suggested that patients should be relatively fit and without serious comorbidities, willing to travel to a specialist centre for CAR T-cell therapy if needed, and have an appropriate home support network, including care support for the acute period (30 days) after CAR T-cell infusion when the risks of neurological adverse events are highest.10,13
Lymphapheresis is a critical step in obtaining starting material for a high-quality CAR T-cell product; therapy should be halted in a timely fashion before apheresis to maximise chances of adequate CD3 yield. A target recommended minimum blood volume of roughly 10–20 L is processed to deliver a final volume of apheresis of approximately 100–500 mL, with a target of approximately 5–10×109 mononuclear cells.5,8,14
Duration and time since last exposure to prior therapies that may impact lymphocyte count should be considered before apheresis (i.e., checkpoint inhibitors, ibrutinib, chemotherapy, and radiotherapy), and use of corticosteroids must also be considered because of their potential lymphotoxicity. Systemic chemotherapy, radiation therapy, and immunosuppressive therapy should be halted in advance of apheresis. At least 2 weeks or 5 half-lives, whichever is shorter, must have elapsed since any prior systemic therapy at the time the patient is planned for leukapheresis, except for systemic inhibitory/stimulatory immune checkpoint therapy.8 Corticosteroids should be avoided 7 days before apheresis. Other medicines with systemic inhibitory/stimulatory immune activities, such as checkpoint molecule therapy, should be stopped approximately 3 half-lives in advance. Intrathecal therapy should be eluded 1 week in advance.8,14 Patient blood composition can impact lymphocyte yield in the apheresis; high proportions of natural killer cells or myeloblasts, and low absolute lymphocyte count in peripheral blood, can compromise CD3 yield and lead to a low-target apheresis harvest; low CD3+ T-cell yield can lead to impaired CAR T-cell expansion in vitro.15 Although the percentage of T cells in the starting materials may vary widely, with the result being being that some patients were essentially lymphopenic, the resulting T-cell products were highly enriched for CD3+ T cells in ZUMA-1.16
Shipping via cold chain is the final piece of the process; on the day of apheresis, Kite provides a NanoCool® container for shipping. Apheresis material is collected from hospital laboratories and transported to one of Kite’s manufacturing locations for peripheral blood mononuclear processing.17 The KiteKonnect® online portal provides a range of supporting features to hospitals and healthcare providers to help facilitate the treatment and manufacturing process, including patient enrolment, co-ordination calls, planning of apheresis, and product tracking.
The chain of identity and chain of custody (tracking of one patient, one apheresis material) are critical when treating patients with autologous cell therapy products, and excellent communication between the manufacturer and the treating centre is essential. In ZUMA-1 , median turnaround time was 17 days; therefore, bridging therapy was not allowed to enable a better assessment of the efficacy results.9 However, bridging therapy may be needed between apheresis and CAR T-cell therapy infusion whilst patients are awaiting the processed cells, as has been demonstrated in more than half of patients in the real world in the USA.12,18 Patient management should be collaborative between the CAR T-cell treatment and referring centres; preferred therapies are those that will not make patients unwell or negatively impact their performance status. Consensus guidance around standardisation of apheresis processes may help to provide uniformity of CAR T-cell products across treating centres and manufacturers.
From Sample Receipt to CAR T-cell Product
Doctor Chris Shen and Mister Louis van de Wiel
The axicabtagene ciloleucel product flow is an aligned process between the manufacturer, the apheresis unit, and the treating hospital to return the product to the patient timely. Teamwork is vital and an efficient manufacturing process requires seamless integration and communication between multiple stakeholders, including physicians and Kite internal teams. The flow starts at the Kite manufacturing facility with generation of the chain of identity and then proceeds with shipping of the apheresis materials kit to the relevant apheresis unit, where the chain of custody starts. After the cells are collected, they will be shipped to the manufacturing facility and inspected for quality and condition before manufacturing begins. QC testing is also a vital part of all the steps in the production process.16
In ZUMA-1 at the USA sites, the manufacturing success rate was 99% with a median turnaround time of 17–18 days.9 In Europe, the turnaround ranges from 26 to 29 days, mainly due to the transport time needed between Europe and the USA for cell processing.19 The USA real-world manufacturing success rate is 97%.12 Kite is based in California, with research and development, and commercial and clinical manufacturing facilities in the Los Angeles area in the USA. Kite also has entered into joint ventures/license collaborations with companies in Japan (Daiichi Sankyo)20 and China (Fosun Kite).21 Kite is committed to increasing patient access in Europe, and manufacturing network expansion is underway. A new facility in the Amsterdam area of the Netherlands will be operational by the beginning of 2020 and will supply the European market, with the capacity to produce approximately 4,000 patient treatments per year.22
The CAR T-cell therapy process begins with apheresis, which can be highly variable depending on patient status and collection site. This step is followed by T-cell isolation and enrichment. As patient materials can have variable volume and cell concentrations, volume normalisation may be performed as the first step to ‘standardise’ starting material. The difference in manufacturing time between the USA and the EU is due to the current EU need for peripheral blood mononuclear cells to be cryopreserved and shipped to the USA. T cells are then activated to provide the primary signal; this is then followed by gene transduction of T cells (engineering with CAR). Anti-CD19 CAR genes are introduced into cells by retroviral transduction.16 CAR T-cell expansion then follows. A single dose of axicabtagene ciloleucel contains a target dose of 2×106 CAR-positive viable T cells per kg of body weight (range: 1×106–2×106 cells/kg; up to a maximum of 2×108 CAR-positive viable T cells for patients ≥100 kg) in approximately 68 mL dispersion in an infusion bag.10
T-cell activation, transduction, growth, and final formulation are all critical to an efficient manufacturing process. Expansion is then followed by QC testing.5,16 Cell growth rates can be variable depending on the starting product (the growth process takes 4–7 days), but the manufacturing process is designed to accommodate this.
Kite has developed a robust and efficient approach to engineered cell therapy using the patient’s own T cells, and a process enabling consistent, timely delivery of a high-quality product. The future will involve continuing process development with an emphasis on automation, product consistency, increased efficiency and capacity, and decreased manufacturing turnaround time.
Product Release Tests: Helping Ensure Patient Safety and Guaranteeing Drug Product Quality
Professor Christian Chabannon
There are essential differences between the CAR T-cell manufacturing process and the production of conventional drug products. Traditional manufacture of drugs begins with fully characterised and batched starting materials. It is large-scale and mostly automated, resulting in thousands of similar lots for release and distribution. CAR T-cell manufacturing, by contrast, is individualised to each patient (batch size of one), has an inherent variability of starting product, a complex manufacturing process (which is currently mostly manual), uses a specialised facility with highly trained personnel, and rarely encompasses OOS production with exceptional release.23-25 As CAR T-cell manufacturing begins with heterogeneous starting material, release tests and QC of all products are vital to ensure its safety and effectiveness.26 After the CAR T-cell manufacturing process in the USA, the cell product is passed to QC for testing, which comes out 7 days after formulation, and the product is then cryopreserved and shipped back to the European site for batch release and treatment. Before release, a qualified person ensures that each manufactured product batch meets the specifications required by the European Commission.25
During axicabtagene ciloleucel production, each CAR T-cell product undergoes a series of batch-release tests for viability, purity, potency, microbiological safety, appearance, and identity.5,16 Lot release testing serves as the final confirmation of product quality before release for use; these specifications ensure a consistent final product.17 Potency assays are an essential component of release testing. Specifications are agreed upon with competent regulatory authorities during the marketing authorisation process; CAR T-cell potency assays should demonstrate CD19 antigen recognition, T-cell effector function and activation, and be representative of the product mechanism of action.
Increased manufacturing capacity within the new Kite European facility in 202022 should result in removal of the need to ship to the USA and back and, in the best case scenario, align the EU manufacturing time in a similar fashion fashion to that seen in the USA (17–18 days). There are challenges associated with autologous cell therapy manufacturing; it is a specialised process with inherent variability. In real-world experience in the USA, around 3% of the cases received a nonconformity product in the context of ZUMA-9.12 These products are designated OOS.23,24 All medicinal products should be manufactured in compliance with GMP, according to the guidelines for advanced therapy medicinal products (ATMP)-specific GMP published in 2017.23 The Committee of Advanced Therapies (CAT) has recently issued a ‘questions and answers’ document on the use of OOS batches of authorised ATMP.24 OOS products can still exceptionally be administered to avoid an immediate, significant hazard to the patient. This release is dependent on physician request, after which the manufacturer follows a set procedure based on ATMP-specific guidelines.25 If the OOS product is not appropriate to be released exceptionally, then the manufacturing process can be restarted, sometimes even with a new apheresis procedure which may require additional management of patient disease during this extra period.
The individualised nature of CAR T-cell therapy introduces inherent variability at the start of the manufacturing process.27 Apheresis variability may be among the causes of product variability.15 QC from the beginning to the end of the manufacturing processes of all CAR T-cell therapies is essential to ensure the safety and effectiveness of these products.26
In conclusion, the axicabtagene ciloleucel manufacturing process is robust, with success rates of 99% in ZUMA-18 and 97% in the real-world setting in the USA.12 Although the majority of manufacturing events are successful, manufacturers should continue to improve their processes to minimise OOS production, and implement procedures for risk assessment and communication to the sites of the eventual OOS cases.24
Scaling CAR T-Cell Therapy for a Good Manufacturing Practice Standard Product
Doctor Claire Roddie
The CAR T-cell infusion process starts with patient admission for lymphodepleting chemotherapy. A conditioning regimen of fludarabine (30 mg/m2) and cyclophosphamide (500 mg/m2) is used for standard lymphodepletion (over 3 days from Day5 before CAR T-cell infusion; three doses of fludarabine and three doses of cyclophosphamide). This step in patient conditioning is essential for depleting endogenous lymphocytes and elevating homeostatic cytokines (i.e., IL-15 and IL-7).4 The blood bank and the patient should be informed of the requirement for irradiated blood products, and the treating physician should ensure that at least 2 days elapse between the last dose of lymphodepleting chemotherapy and CAR T-cell administration (to ensure the clearance of fludarabine from the system). All patients should be made aware of the risks of neutropenia and possibly sepsis with this conditioning regimen.10
At the time of infusion, hospitals should ensure the existence of checklists to cover all the processes. Patient identity must be checked before removing the bag from the cassette. Then, the bag must be thawed at 37 °C either in a water bath or using dry methods for 3–5 minutes. No further manipulation of the product (e.g., washing, resuspension, or radiation) is permitted. As required with other infusions of lymphocytes or haemopoietic progenitors, a leukodepleting filter must not be used.
During the postinfusion period, patients must be closely monitored for 4 hours for infusion reactions. Tocilizumab must be immediately available in case it is needed for management of CRS, which occurs within 1–12 days. Thus, patients must be monitored in a treatment facility for 10 days after infusion. The summary of product characteristics and the additional risk minimisation materials approved by competent national authorities provides guidelines for management of CRS and neurological toxicity.10
Why do we Need to Scale Out for a Good Manufacturing Practice Standard Product?
The CAR T-cell journey includes many steps such as apheresis, T-cell enrichment, gene transduction, T-cell expansion, formulation, and testing. Current commercialised CAR T-cell products are individualised to each patient and require a batch per patient, and therefore have a very different manufacturing process than that used in conventional drug manufacturing. ‘Scaling out’ of current CAR T-cell manufacture requires increased labour, materials, equipment, and space.28 Challenges in scaling out include ensuring that CAR T cells are manufactured according to GMP requirements, a system by which medicinal products are produced safely according to standardised methods under tightly controlled, reproducible, and auditable conditions. Fundamental quality systems required in a GMP facility include items described in Figure 2.15
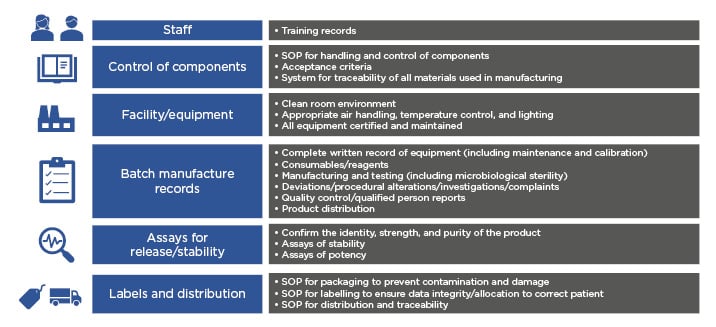
Figure 2: Scaling-out quality systems required of a current good manufacturing practice facility.
SOP: standard operating procedures.
Adapted from Roddie C et. al.15
In the future, some of the current logistical challenges may be improved at different points of the process: eliminating the cool chain manufacturing requirement, harmonising national cell transport license variations, enhancing and simplifying digital tracking of individual identity of patient cells, implementing standardised global release procedures, and potentially identifying and removing unnecessary tests.
Cell therapy manufacturing is proceeding to overcome these challenges, with manufacturing capacity being scaled up, increased use of automation, highly trained staff, samples being tested and quality assured, site qualification, improved transport and logistics, and, importantly, maintenance of the chain of custody. Kite is currently deploying a standardised process of qualification of all the treatment sites, including steps such as audit, training in the additional risk minimisation materials, and a dry-run exercise to ensure high-quality treatment.17
Currently, autologous CAR manufacturing involves scaling out to produce more therapies, with replication of the same scale of equipment and processes performed in paralle. In the future, scale-up as per conventional treatments may involve an increase in batch size with ‘batched’ products from a single allogeneic source (a healthy source where multiple batches can be made from that single donor).16 Alternatives to viral transduction exist, which may remove one huge hurdle to CAR T-cell manufacture: the production of GMP-ready viral vectors to manufacture these cells. Thus, allogeneic production could remove a lot of the complexity involved in the current autologous manufacture system.
In conclusion, the scaling out of autologous CAR manufacture to meet the needs of many patients in DLBCL has been shown to be feasible. The current manufacturing process is continuously undergoing improvements. Further automation is being investigated, which may give more benefit to overcome some of the challenges and shorten the turnaround time. Scale-out has already been successfully achieved with axicabtagene ciloleuce in more than 1,000 patients in clinical trials and in real-world treatment.10,12,18





