
Characterisation of β-Thalassaemias
Doctor Clara Camaschella and Professor Ali T. Taher
β-thalassaemias are inherited blood disorders caused by a defect in the production of β-globin, giving rise to variable phenotypes, ranging from severe anaemia to an asymptomatic phenotype.1 The severity of the mutation on each β-globin allele is classified as β0, a complete absence of β-globin on the affected allele, β+, a residual production of β-globin (from 10–50%) or β++, a very mild reduction in β-globin production.1 β-thalassaemia is classified as β-thalassaemia major, β-thalassaemia intermedia, and β-thalassaemia minor.1
More recently, thalassaemias have been classified by their requirement for transfusion into non-transfusion -dependent thalassaemias (NTDTs) and transfusion- dependent thalassaemias (TDTs) (Figure 1). Patients with NTDTs do not require regular transfusion for survival; however, they may require occasional transfusion due to growth failure, pregnancy, infections, and other specific situations.1-5
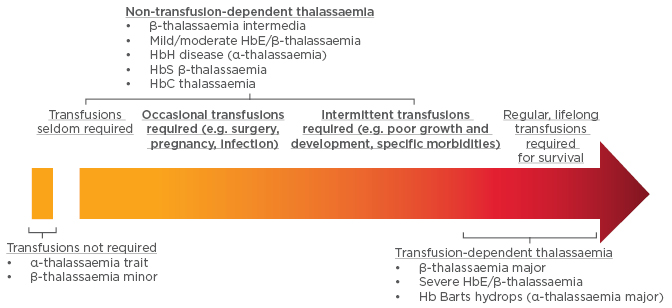
Figure 1: Designation of thalassaemias based on transfusion requirements.
Hb: haemoglobin.
Adapted from Musallam et al.5
The prevalence of NTDT is increasing worldwide as increased migration flow spreads the reach of these diseases.6 β-thalassaemia intermedia is most common in Africa, the Mediterranean, India, and Eastern Europe;7,8 HbH/α-thalassaemia is most common in South East Asia, the Middle East, and the Mediterranean;6,9 and HbE/β-thalassaemia is most common in East India, Bangladesh, and South-East Asia.7,9
The severity of β-thalassemia is very much dependent on the free alpha chains not paired to beta, leading to ineffective erythropoiesis. Numerous studies of the genetic determinants of disease severity in β-thalassaemia have demonstrated that the symptoms of β-thalassaemia can be ameliorated by an increased or persistent production of ɣ-globin. In patients where there is an incomplete switch from the production of ɣ-globin to β-globin, the ɣ-globin has been shown to mop up the free α-globin chains and considerably ameliorate the phenotype of β-thalassaemia. Co-inheritance of α-thalassaemia and β-thalassaemia can also reduce symptoms, due to the down synthesis of α-globin resulting in a smaller pool of free α-globin chains.
Normal Erythropoiesis and Disruption of Pathways in β-Thalassaemia
Doctor Clara Camaschella
Steady-state erythropoiesis involves the process of proliferation and differentiation of new red blood cells from erythroid progenitors, which primarily occurs in the bone marrow.10-14 Erythropoiesis is regulated by erythropoietin (EPO), which is released from the kidney in response to hypoxia and hypoxia-inducible factors. EPO binds to EPO receptors on the membrane of the progenitor erythroid cell, activating gene expression via the Janus kinase signal transducer and activator of transcription 5 pathway (JAK2/STAT5) (Figure 2). Another essential erythroid transcription factor is GATA1 (Figure 2).
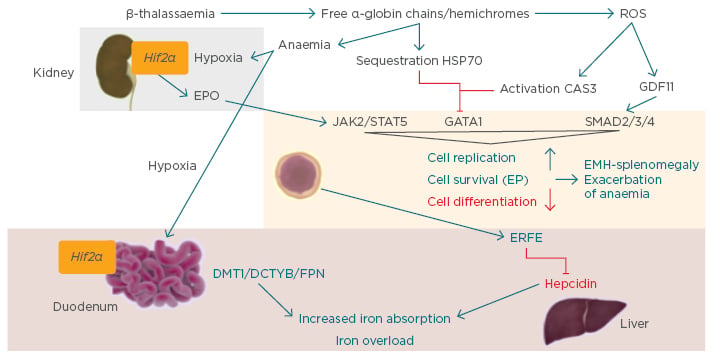
Figure 2: Ineffective erythropoiesis.10
CAS3: caspase 3; DCYTB: duodenal cytochrome b; DMT1: divalent metal transporter 1; EMH: extramedullary haematopoiesis; EP: erythroid progenitor; EPO: erythropoietin; ERFE: erythroferrone; FPN: ferroportin; GATA1: GATA binding protein 1; GDF11: growth differentiation factor 11; Hif2α: hypoxia-inducible factor 2α; JAK2: Janus kinase 2; ROS: reactive oxygen species; STAT5: signal transducer and activator of transcription 5.
Stimulation of GDF11, a ligand of the activin receptor (ACVR)2B, has also been demonstrated to control differentiation of erythroid cells through the inhibitory SMAD2/3/4 pathway. Together these mechanisms control the cell replication, survival, and differentiation of the erythroid progenitors. Increased EPO production or JAK2 pathway mutations may lead to erythrocytosis, whilst conditions of EPO deficiency, ineffective erythropoiesis, or inflammation can result in anaemia.
The main phenotypical characteristics of β-thalassaemia are the result of production of a relative excess of α-globin chains and the production of free α-globin chains. These excess α-globin chains precipitate in red blood cells and immature erythroid cells causing premature destruction of erythroid precursors (ineffective erythropoiesis). An excess of α-globin chains causes the majority of defects in the erythropoietic pathway as they aggregate in hemichromes, and lead to the vicious cycle of anaemia and hypoxia that eventually result in overproduction of EPO in the kidney. EPO activates the JAK2/STAT pathway in the erythroid progenitor cells. In parallel, the free α-globin chains are sequestered by heat shock protein (HSP)70, a chaperone protein, which protects the GATA1 from caspase 3 cleavage in the nucleus. HSP70 engaged by free α-chains cannot translocate to the nucleus and protect GATA1.15 Hemichromes generate reactive oxygen species, which activate caspase 3 and inhibit GATA1 while activating GDF11 and the inhibitory pathway SMAD2/3/4. These mechanisms together increase cell replication and survival of progenitors and reduce differentiation of the cells, leading to ineffective and extramedullary haematopoiesis (EMH)-splenomegaly and exacerbation of anaemia (Figure 2).
Iron Homeostasis in β-Thalassaemias
Doctor Clara Camaschella, Professor Ali T. Taher, and Doctor Maria-Domenica Cappellini
The conventional treatment of severe β-thalassaemias is regular blood transfusion, which is associated with iron overloading and results in multiple extrahepatic morbidities. To prevent organ complications due to iron overload the patient must be chelated,5,16,17 as the human body has many mechanisms for absorbing, transferring, and storing iron, but none for its excretion.18 In addition to an increase in EPO, ineffective erythropoiesis in β-thalassaemia leads to increased production of erythroferrone. Erythroferrone, a peptide hormone synthesised primarily in the liver and responsive to ineffective erythropoiesis, has recently been shown to inhibit hepcidin. Hepcidin is the main circulating regulator of iron absorption and tissue distribution.19,20 Hepcidin regulates dietary iron absorption and distribution by triggering the degradation of ferroportin-1, the membrane-bound iron exporter found in enterocytes, hepatocytes, and macrophages. Hepcidin is upregulated by increased iron levels and inflammation, and downregulated in conditions associated with accelerated erythropoiesis such as anaemia and hypoxia.19,20 The combined effect of the increase in EPO and reduction of hepcidin leads to increased intestinal iron absorption and an increased release of recycled iron from the reticuloendothelial system, resulting in iron overload and toxicity in several organs.16
The most common test for iron overload is serum ferritin; however, liver iron concentration remains a gold standard, as the liver is the primary site for iron storage (>70% of body iron) and correlates with the total body iron.21 Given that liver iron concentrations ≥5 mg/kg have been linked to an increased prevalence of vascular, endocrine, and bone morbidities, the latest Thalassaemia International Federation (TIF) guidelines advise that, as a minimum, serum ferritin should be tested every 3 months, magnetic resonance imaging (MRI) to measure iron liver concentration should be measured every year and T2* MRI of the heart should be undertaken every 1–2 years, according to the patient’s iron status.17,22 Observational data demonstrate that serum ferritin levels may underestimate the extent of iron overload in thalassaemia intermedia and therefore should not be used as a negative predictor of significant iron overload in this patient population, resulting in prolonged patient exposure to high iron levels and the associated risk of morbidity and mortality.23-25 As β-thalassaemia is most prevalent in developing countries, recommendations for iron monitoring must also consider the availability of resources. To allow for scenarios where MRI facilities are unavailable, a clinical scale indicating the complications associated with liver iron concentration, measured by MRI or liver biopsy, has been developed (Figure 3).17,26
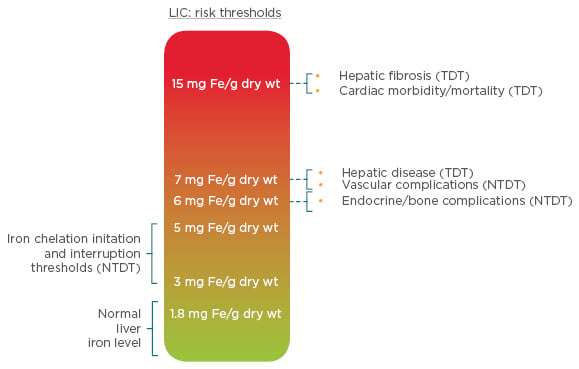
Figure 3: Liver iron concentration thresholds and their clinical implications in NTDT.
LIC: liver iron concentration; NTDT: non-transfusion-dependent thalassemia; TDT: transfusion-dependent thalassaemia; wt: weight.
Adapted from Taher et al.17
Iron chelation therapy aims to maintain safe levels of body iron by balancing iron intake with iron excretion, to remove excess stored iron that has accumulated after blood transfusions, or in emergencies as an intense treatment to remove excess iron quickly to reverse the effects of heart failure.26 Iron chelation therapy for thalassaemia patients is evolving; in the 1970s regular transfusion was normal practice, which progressed to chelating agents in the 1980s with the introduction of deferoxamine (DFO). In 1999, the T2* MRI technique was introduced in the UK, and in the same year deferiprone (DFP) was approved in Europe, followed by approval of deferasirox (DFX) in 2006; both are oral drugs whereas DFO is administered by subcutaneous infusion. Parallel to these advances in chelation therapy, a reduction in mortality was observed from 12.7 deaths per 1,000 patient years from 1980–1999, to 4.3 deaths per 1,000 patient years from 2000–2003.27 Use of DFO has reduced significantly since the introduction of the oral iron chelators DFP and DFX; however, the use of subcutaneous/intravenous DFO is still utilised for patients with heart failure or when a quick reduction of elevated serum ferritin is required (7,000–10,000 ng/mL). Combination therapy of DFO and DFP can also enhance myocardial iron removal in TDT patients with myocardial overload.28 DFX provides significant improvement in iron overload according to myocardial T2* MRI over a 3-year period in patients with TDT, and liver iron concentration and serum ferritin were halved in severely iron-overloaded patients over the same time period.29 These improvements are accompanied by an improvement of liver pathology status in patients with β-thalassaemia.30
Clinical Considerations for Non-Transfusion-Dependent Thalassaemias
Professor Ali T. Taher
Even patients who are not transfusion dependent may require transfusions in the event of surgery, infection, or during the course of pregnancy.5 In children with severe growth retardation, it is sensible to transfuse over a period of time until the child has normalised growth. In addition, populations at risk of thrombosis may be transfused regularly as a preventive measure. Although patients with NTDT present with a milder anaemia than patients with TDT do, they are still at risk for serious and often irreversible morbidity as they advance in age.5,16 For NTDT patients, occasional or regular transfusion may be protective against a number of complications, including thrombosis, EMH, pulmonary hypertension, heart failure, cholelithiasis, and leg ulcers; however, transfusion therapy is associated with an increased risk of endocrinopathy.31 Consideration should also be made regarding the risk of alloimmunisation, particularly in older or pregnant patients.32
Enlarged spleen (splenomegaly) and overactive spleen (hypersplenism) are inevitable in untransfused or rarely transfused NTDT patients due to EMH, splenic pooling, and chronic passive congestion of the spleen.2,33 Hypersplenism often leads to worsening anaemia, neutropenia, and thrombocytopenia, and splenomegaly can be associated with abdominal pain and risk of splenic rupture.2,33 Splenectomy is performed in rare cases in NTDT patients as it can increase the total haemoglobin level by 1–2 g/dL and avoid the requirement for blood transfusions.2 Cholecystectomy during splenectomy may also be used in NTDT patients who are at increased risk of gall stone formation from increased haemolysis.2 Splenectomy is associated with higher incidences of infection and vascular abnormalities, such as venous thromboembolism, pulmonary hypertension, and silent cerebral infarction.2,33 Splenectomy is generally avoided in NTDT patients younger than 5 years and is reserved for:17
- Cases of worsening anaemia leading to poor growth and development when transfusion therapy is not possible or iron chelation therapy is unavailable
- Hypersplenism where leukopenia or thrombocytopenia are causing clinical problems such as recurrent bacterial infection or bleeding
- Splenomegaly accompanied by symptoms such as left upper quadrant pain or early satiety or massive splenomegaly (largest dimension >20 cm) with concern about possible splenic rupture
In the absence of transfusion, ineffective erythropoiesis may lead to a variety of subsequent sequelae, including marrow expansion and decreased bone mass as well as the development of extramedullary pseudotumours that can exert pressure on anatomical structures such as the spinal cord. Although EMH is a rare condition, it is more common in this group of patients.34 In patients not receiving regular transfusions, there is increased bone marrow activity to overcome the anaemia and apparently normal blood cells form outside the bone marrow.34 Formation of erythropoietic tissue masses occurs mainly in the chest area and near the spine, but these may also affect the spleen, liver, and lymph nodes.35-38 Symptoms develop as a result of pressure on the surrounding structures, and spinal cord compression may cause possible irreversible damage.35-38 MRI is the best diagnostic method in these cases.34 Clinical management of EMH includes transfusion therapy, radiotherapy, hydroxyurea, or surgery.34
Patients with NTDT also possess a hypercoagulable state that can lead to vascular complications, including thrombosis, pulmonary hypertension, and leg ulcers, with greater risk in those patients who have had a splenectomy.39 Patients who have undergone splenectomy have higher incidence of pulmonary hypertension, and increased liver iron concentration and advancing age have also been demonstrated to correlate with pulmonary hypertension.40-42 A patient with NTDT should be considered a medically ill patient and they should be evaluated in a risk assessment model to determine if they are at higher risk of thrombosis, and whether they should be treated prophylactically for surgery or during pregnancy, and if they should be educated about the use of contraceptives. Further clinical complications associated with iron overload in NTDT include hepatic fibrosis, cirrhosis and cancer, endocrinopathies, and osteoporosis.5
New Treatment Targets in Thalassaemias
Doctor Maria-Domenica Cappellini and Doctor Clara Camaschella
Treatments for Iron Chelation and Iron Overload
Conventional therapy in β-thalassaemia focusses on targeting ineffective erythropoiesis due to blood transfusions, iron overload, or anaemia.5 In the iron chelation field, the DFX dispersible tablet provided great improvements in DFO and DFP, but with some limitations due to reduced treatment adherence as a result of palatability and preparation issues.43,44 A new formulation of DFX has been developed as a once-daily film-coated tablet (FCT), without lactose, which can be taken with a light meal.44,45 Significant improvements in adherence, patient satisfaction, and patient preference were observed for the FCT in the ECLIPSE study, an open-label randomised, multicentre, two arm Phase II trial comparing DFX dispersible tablet with DFX FCT.46,47
Iron overload can also be managed by targeting the iron homeostasis pathway. Hepcidin is proposed as a target for β-thalassaemia based on the hypothesis that it can block the accumulation of excess iron in erythroid cells, thereby decreasing iron overload and the formation of toxic hemichromes, and ameliorating erythroid parameters leading to both a reduction of ineffective erythropoiesis and an increased lifespan of red blood cells. Preclinical studies show that increased hepcidin expression limits iron overload, with an associated improvement in haemoglobin levels because of increased red blood cell survival and reduced ineffective erythropoiesis and splenomegaly.48-50 Minihepcidins are short peptide mimetics (9-retro-inverso amino acid) of hepcidin (25 amino acid) that have been shown to be effective in reducing iron overload in animal models of β-thalassaemia.51,52 In animal models of β-thalassaemia intermedia, minihepcidins improve ineffective erythropoiesis, splenomegaly, iron overload, red blood cell lifespan, and anaemia. Similarly, in β-thalassaemia major models they demonstrate significant amelioration of anaemia and splenomegaly, and enhance the transfusion-mediated effect of suppressing ineffective erythropoiesis and reversing splenomegaly. The effects seemed to be maintained when minihepcidins were associated with iron chelation.
Treatments to Change the Genetic Imbalance of α and β-globin
Considering that the imbalance between α and β chains is the driver of the pathophysiology of β-thalassaemia, targeting the α and β chain imbalance or targeting ineffective erythropoiesis, which is the consequence of the imbalance, are very interesting options for the treatment of β-thalassaemia. There are two known methods to control this imbalance used in clinical practice: bone marrow transplantation and gene therapy.
The recommendations for bone marrow transplantation in thalassaemia and sickle cell disease were published in 2014 and will be updated soon.53 Optimal bone marrow transplantation in thalassaemia is achieved with human leukocyte antigen-identical donors, as alternative donors are associated with less favourable outcomes. It has been suggested that human leukocyte antigen-haploidentical stem cell transplantation following removal of T and B cells demonstrates positive results in children with non-malignant disorders, and therefore bone marrow transplantation in β-thalassaemia is now an expanding area of possibility.54 Although it might seem a paradox, bone marrow transplantation continues to be an important treatment option for countries with limited resources, because it is a curative option.
One of the most exciting areas of β-thalassaemia therapy research is in the field of gene therapy. Gene therapy has been proposed as a potential treatment for β-thalassaemia for >20 years; however, only recently has progress really begun. The objective of gene therapy is to correct the β-globin gene defect to reduce the α/β imbalance in erythroid cells, thereby increasing the normal red blood cell count and reducing ineffective erythropoiesis, leading to transfusion independence. The most difficult phase in the development of gene therapy was the identification of an appropriate vector. The vector should be highly efficient and permit stable transduction, effectively targeting the haematopoietic stem cells, and the transgene expression must be controlled and sustained. In addition, genome toxicity should be absent or low. The lentiviral vector appears to be the most safe, effective, and convenient vector for globin gene expression.
There are two parallel projects ongoing for correction of the beta gene defect by the introduction of a normal beta gene in β-thalassaemia, one supported by bluebird bio® (Cambridge, Massachusetts, USA) and the second from The San Raffaele Telethon Institute for Gene Therapy (SR-Tiget) in Milan, Italy. bluebird bio® selected the vector BB305, which is now in use in clinical practice, whereas the vector validated by SR-Tiget is a highly efficient globe vector.55 bluebird bio® have two ongoing trials in transfusion-dependent β-thalassaemia: a Phase I/II multicentre global study with 18 subjects, and a Phase I/II single-centre study in France with seven subjects (four subjects with β-thalassaemia and three subjects with sickle cell disease). bluebird bio® demonstrated an extended transfusion independence in subjects with mild genotypes (βE/β0 or β+/β+-thalassaemia), with red blood cell independence extending up to 16.4 months; however, no significant improvement was observed for β0/β0-thalassaemia subjects. The next steps for the bluebird bio® gene therapy are two Phase III trials: a Phase III multicentre global study in subjects with transfusion-dependent non-β0/β0-thalassaemia (15 adult subjects and 8 paediatric subjects), which launched in September 2016, and a second Phase III study in β0/β0-thalassaemia (15 adult, adolescent, and paediatric subjects) planned for 2017.
Approval was gained for a trial with the globe lentiviral vector in Milan in three consecutive age groups for gene therapy, including three adult subjects, three adolescent subjects, and four paediatric subjects (TIGET-BTHAL study). Only following results from the adult subjects is treatment permitted in the adolescent group, and only after completion of treatment in the adolescent subjects can the paediatric subjects be enrolled. Preliminary results from the trial suggest a good tolerability of the procedure in adult patients, including conditioning, good tolerability of the intra-bone infusion, and polyclonal engraftment without clonal dominance.
The final pathway to genetically control ineffective erythropoiesis under evaluation in in vitro and preclinical models is genome editing. Successful reactivation of fetal haemoglobin has been achieved by targeting BCL11A through genome editing.56-60 BCL11A is involved in the suppression of fetal haemoglobin, and a developmental stage-specific, lineage-restricted enhancer controls expression of BCL11A in erythroid cells. Therefore, it might be possible to delete BCL11A or its specific erythroid enhancer by genome editing, and prevent BCL11A expression in haematopoietic stem cells or erythroid cells. However, these studies are in their initial phases.
Novel Therapies
New treatments for β-thalassaemia can also target ineffective erythropoiesis through pharmacological intervention, such as disruption of the JAK2/STAT5 pathway with the JAK2 inhibitor, ruxolitinib. High levels of EPO lead to hyperactivation of phospho-JAK2, which expands immature red cell precursors unable to mature and reduce red blood cell production.10 Ruxolitinib is a potent and selective oral JAK1 and JAK2 inhibitor, approved for use in myelofibrosis and polycythaemia vera. Ruxolitinib is associated with a clinically relevant improvement in splenomegaly in these disorders. The efficacy and safety of ruxolitinib was tested in a Phase IIa trial of regularly transfused β-thalassaemia patients.61 The primary endpoint was the reduction of transfusion requirement and the secondary endpoints included reduction of spleen volume.61 Clinically meaningful reductions in mean spleen volume were observed (19.7% at Week 12 [n=26] and 26.8% at Week 30 [n=25]), suggesting that ruxolitinib might serve as an alternative option in patients with TDT who are potential candidates for splenectomy.61
Sotatercept (ACE-011) and luspatercept (ACE-536) act as ligand traps to inhibit ACVR2A and ACVR2B, respectively, thereby inhibiting SMAD 2/3 signalling.62 Luspatercept is currently being investigated in the Phase III BELIEVE study, a double-blind, placebo-controlled trial in TDT with 300 patients enrolled in order to improve haemoglobin levels in NTDT and to reduce transfusion-dependence in thalassaemia major.
Summary
β-thalassaemias include a spectrum of complex clinical phenotypes with varying therapeutic needs. The current treatment paradigm therefore must be tailored to the individual patient’s needs. We are entering a new era of novel therapeutics for β-thalassaemia care, and several targets have been identified that can ameliorate the genetic defect, ineffective erythropoiesis, or iron dysregulation to reduce or prevent β-thalassaemia symptoms.





