
Abstract
Thalassaemia is a hereditary cause of hypochromic microcytic anaemia resulting from defects in haemoglobin production. β-thalassaemia, which is caused by a decrease in the production of β-globin chains, affects multiple organs and is associated with considerable morbidity and mortality. This review aims to highlight the significant progress being made in the areas of ineffective erythropoiesis control, metal chelation, and gene therapy, which is bringing new hope and should change patient management and prognosis in the near future.
INTRODUCTION
Thalassaemia is a hereditary cause of hypochromic microcytic anaemia resulting from defects in haemoglobin (Hb) production.1 Hb is the iron-containing oxygen-transport metalloprotein in red blood cells (RBC). Its synthesis is controlled by two multigene clusters on chromosome 11 (β-globin) and chromosome 16 (α-globin). A Hb molecule comprises four globular protein subunits; each subunit is composed of a protein chain tightly associated with a non-protein prosthetic haem group which consists of an iron ion (Fe2+) held in a heterocyclic ring, known as a porphyrin. The adult Hb (HbA) that is composed of two α chains and two β chains (α2β2) represents almost all Hb in adults (95.5–97.0%). HbA2, which is composed of two α chains and two δ chains (α2δ2), represents 2.0–3.5% of HbA. Fetal Hb is composed of two α chains and two γ chains (α2γ2) and represents 1.0% of HbA. When a decrease in synthesis affects β chains, a compensatory mechanism leads to an increased elaboration of γ and δ chains, hence an increase of HbF and HbA2. β-thalassaemia, which is caused by a decrease in the production of β-globin chains, affects multiple organs and is associated with considerable morbidity and mortality. The highest prevalence of β-thalassaemia mutations is in people of Mediterranean, Middle Eastern, and Asian descent. Over 200 different thalassaemia-causing mutations have been identified in the β-globin gene, leading to wide genotypic and phenotypic variability of the disease.2 There are several types of mutations: silent mutations (silent β), mild mutations that cause a relative reduction in β-globin chain production (β+), severe mutations that result in complete absence of β-globin chain synthesis (β0), and the most uncommon, deletion of the gene.3 β-thalassaemia minor is a heterozygous inheritance of a β-thalassaemia mutation, with patients often having clinically asymptomatic-microcytic anaemia. Patients with β-thalassaemia major, with homozygous mutations, usually present with severe anaemia in infancy and become transfusion-dependent for life, whereas patients with β-thalassaemia intermedia present later in life with mild-to-moderate anaemia and variable transfusion requirements.2 Both β-thalassaemia major and intermedia can result from the homozygous or compound-heterozygous inheritance of mutations in the β-globin gene. Several modifications can result in patients having β-thalassaemia intermedia rather than major, including ineffectiveness of erythropoiesis or an excess of α-globin.2 Given the many complications, lifelong care is required.
MICROCYTIC ANAEMIA IN β-THALASSAEMIA
HbA tetramer consists of two α chains and two β chains. A lack of synthesis of β chains therefore leads to an excess of α chains. These α chains can precipitate in the cytoplasm of erythroid precursors during haemoglobinisation at the stage of polychromatophilic erythroblast, instigating a deleterious oxidative cascade.1 During physiological erythropoiesis, the chaperone heat shock protein 70 (HSP70) accumulates in the nucleus of differentiating erythroblasts, protecting the major erythroid transcription factor GATA-1 from caspase-3 cleavage.4 This key apoptotic enzyme has an ambivalent action in erythropoiesis. On the one hand, it is essential for terminal erythroid differentiation by selectively cleaving certain targets, which leads, for example, to the condensation of the erythroblast nucleus and to the morphological changes that recede the enucleation, allowing the formation of the reticulocyte.5 It can, on the other hand, have a deleterious function. In fact, the lack of erythropoietin (EPO) promotes the nuclear release of HSP70, which allows caspase-3 to cleave GATA-1 (which is no longer protected) and blocks erythroblastic maturation.4 The nuclear localisation of HSP70 thus represents a fine mechanism of regulation for erythropoiesis, which is necessary because this process represents an extraordinary proliferation system. In β-thalassaemia, HSP70 is sequestered in the cytoplasm of mature erythroblasts (stage of intense haemoglobinisation) during erythropoiesis to perform its natural chaperoning of α-globin chains, which otherwise form excessively toxic aggregates. This will result in a lack of HSP70 localisation and, as a result, the destruction of GATA-1, causing maturation arrest and cell death. With the bone marrow producing 200 billion RBC every day, it is necessary to very quickly control excess production, which can lead to polycythaemia, or compensate for a defect, which would quickly result in anaemia.6 Microcytosis occurs due to a lack of Hb as a result of defects in either globin or haem synthesis, or iron deficiency, leading acidophilic erythroblasts to enter mitosis once more, reducing the volume of RBC. If, however, the cytoplasm of RBC contains a haemoglobin concentration lower than normal, hypochromia is observed.
IRON OVERLOAD IN β-THALASSAEMIA
Erythropoiesis is governed by erythropoietin (EPO), a hormone primarily produced by the kidney. EPO levels, stimulated by hypoxia, are high in β-thalassaemia. EPO binding to its receptor (EPOR) on the surface of erythroid precursors activates the JAK2/STAT5-signalling pathway and the transcription of several genes involved in proliferation, differentiation, and survival.7-9 Despite high EPO levels caused by the globin defect, erythroid differentiation is blocked in β-thalassaemia and the resulting hallmarks of the disease are ineffective erythropoiesis and anaemia. Because of the expanded erythropoiesis, the increased erythroid regulator erythroferrone suppresses hepcidin and causes iron overload (Figure 1).10,11
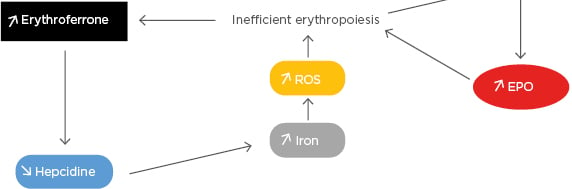
Figure 1: The increased erythroid regulator erythroferrone suppresses hepcidin and causes iron overload.
EPO: erythropoietin; ROS: reactive oxygen species.
NEW INSIGHTS IN THE TREATMENT OF β-THALASSAEMIA
Erythropoiesis-Stimulating Agents
Recombinant human EPO is widely used for the treatment of anaemia, for example, in patients on chemotherapy or on haemodialysis. Treatment with EPO was also tried experimentally in patients with thalassaemia.12,13 In these patients, despite the state of chronic anaemia, EPO levels are usually low relative to the degree of anaemia.14,15 Administration of EPO to splenectomised patients with β-thalassaemia intermedia resulted in a dose-dependent improvement in their anaemia.16 Furthermore, in addition to stimulating RBC and fetal Hb production, EPO might alleviate symptoms of haemolytic anaemias by acting as an antioxidant.17 However, recombinant EPO is not able to correct inefficient erythropoiesis, therefore its use is not recommended for the treatment of β-thalassaemia.
Sotatercept is a ligand trap that inhibits TGF-β superfamily members including growth differentiation factor 11 (GDF-11) and activin B.GDF-11 is overexpressed in immature erythroblasts in β-thalassaemia.18 Aberrant GDF-11 production may induce expansion of erythroid progenitors and increase oxidative stress, leading to maturation arrest of late erythroid precursors and ineffective erythropoiesis.18 Preclinical work has shown that administration of an activin receptor IIA ligand trap decreases GDF-11 concentration, reduces reactive oxidative stress levels, and promotes terminal maturation in immature erythroblasts.18 Sotatercept is a novel recombinant fusion protein consisting of the extracellular domain of the human activin receptor IIA linked to the human immunoglobulin G1 Fc domain19 with interesting efficacy to reduce transfusion requirements in a Phase II trial in myelodysplastic syndromes.20 A recent Phase II trial with 16 patients with transfusion-dependent β-thalassaemia and 30 patients with non-transfusion-dependent β-thalassaemia showed promising results, with most non-transfusion-dependent β-thalassaemia patients treated with high doses achieving sustained increases in haemoglobin level. Transfusion-dependent β-thalassaemia patients who were treated with high doses of sotatercept also achieved reductions in transfusion requirement.21
Iron Overload Targeting Agents
Transfusion and iron chelation therapy can be a lifelong requirement for many patients with β-thalassaemia. Iron enters the plasma and exceeds the capacity for transport by circulating transferrin. Therefore, non-transferrin-bound iron exists in the plasma of thalassaemic patients causing multiple damages when entering cells through the generation of reactive oxygen species. There is a great interest in developing novel treatments that target iron overload by improving erythroid maturation. Iron chelation therapy can influence the frequency and severity of iron overload-related complications, with demonstrated improvement in organ dysfunction and survival in patients with iron chelation therapy.2 Substantial progress has been made to improve the compliance of iron chelating treatments. Initially, the patients had a subcutaneous treatment with deferoxamine at their disposal, before the development of oral treatments such as deferiprone and deferasirox which had a recent galenic evolution.22 Other approaches to decrease iron overload have been tested and shown to have beneficial effects, primarily in models of thalassaemia intermedia (Figure 2). These include restricting iron for erythropoiesis with the use of hepcidin/minihepcidins23,24 or transferrins,25,26 inhibiting the transmembrane protease serine 6, also known as matriptase-2 protease (TMPRSS6),27-30 and targeting Transferrin Receptor 2 (TfR2).31 Defining the molecular mechanism of these novel compounds and exploiting potential combinations is a therapeutic challenge for the future.
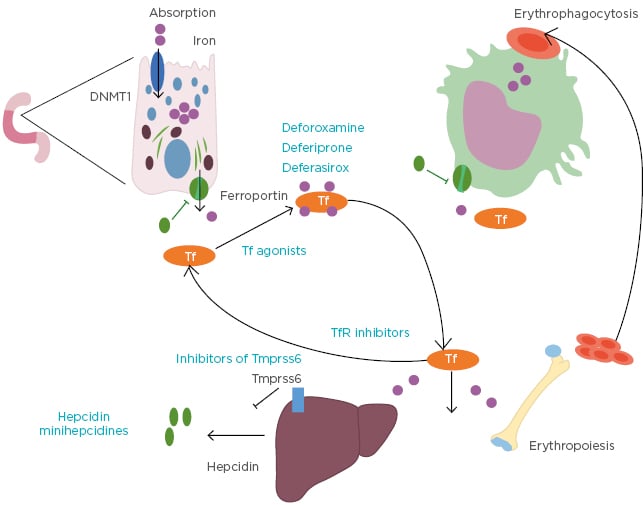
Figure 2: Iron overload-targeting agents.
DNMT1: DNA methyltransferase 1; Tf: transferrin; TfR: transferrin receptor; TMPRSS6: matriptase-2 protease.
Allogeneic Haematopoietic Stem Cell Transplantation in β-thalassaemia
Replacement of mutant RBC using allogeneic haematopoietic stem cell transplantation (allo-HSCT) was, until recently, the only existing curative therapy for β-thalassaemia and is now an established approach to correct inefficient erythropoiesis, particularly when matched sibling donors are available.2 Allo-HSCT is applied with a disease-free survival exceeding 80% with HLA-matched-sibling donors.32,33 Moreover, improvements in conditioning treatment have encouraged the use of alternative donors and umbilical cord blood for the HSC source in instances where patients do not have a matched-sibling donor. Nevertheless, matched-unrelated transplants for high-risk patients with thalassaemia have a long-term overall survival of <70%.34
Gene Therapy in β-thalassaemia
Donor availability and transplantation-related risks have limited the use of allo-HSCT in patients with β-thalassaemia, thus gene therapy is currently evaluated as a new option.35-37 Gene therapy has been shown to hold promise in β-thalassaemia patients using β-globin lentiviral vectors.38,39 In two Phase I–II studies, HSC from 22 patients with transfusion-dependent β-thalassaemia were harvested and transduced ex vivo with LentiGlobin BB305 vector, which encodes HbA with a T87Q amino acid substitution (human βA-T87Q-globin gene). The cells were then reinfused after the patients had undergone myeloablative busulfan conditioning. This allowed the reduction (19 patients) or elimination (3 patients) of the need for long-term RBC transfusions.40 Progress is currently being made regarding the use of lentiviral vectors in terms of transduction rate, but the risk of clonal haematopoiesis persists. Genome editing represents an alternative strategy for the treatment of haemoglobinopathies.41 This is based on the sequence-specific targeting of a nuclease to the genome and the repair of the double-strand break by either non-homologous end joining or homology-directed repair. Non-homologous end joining alters the genome by small insertions or deletions and is induced by targeted double-strand breaks to remove DNA-regulatory elements or to prevent expression of a protein. Therapeutic double-stranded or single-stranded DNA is provided together with a targeting nuclease to mediate homology-directed repair in which a mutant sequence can be replaced by the wild-type sequence. The targeting molecules can either be synthetic DNA-binding proteins like zinc finger (ZF) and transcription activator-like effector (TALE) proteins, or RNA in the context of the CRISPR/Cas9 system.42,43 ZF or TALE proteins are usually fused to the Fok1 nuclease generating ZF-nuclease or TALE-nuclease. Among the three systems, CRISPR/Cas9 has advantages with respect to ease of design and expense. All three systems have limitations including the possibility of generating off-target double-strand breaks and chromosomal rearrangements.43 Current efforts focus on generating nucleases with increased specificity. Genome editing holds great promise for the development of new and globally applicable therapies for haemoglobinopathies, especially β-thalassaemia (Figure 3).

Figure 3: Genome editing strategies.
TALEN: transcription activator-like effector nuclease.
CONCLUSION
β-thalassaemia is still a major public health issue. Significant progress in the areas of ineffective erythropoiesis control, metal chelation, and gene therapy is bringing new hope and should change patient management and prognosis in the near future.





