
Abstract
Although idiopathic aplastic anaemia (AA), myelodysplastic syndrome (MDS), and paroxysmal nocturnal haemoglobinuria (PNH) are all associated with bone marrow failure, they have traditionally been understood as distinct diseases with sharply contrasting pathophysiologies. These three disorders show considerable overlap. In 10% of cases of MDS the bone marrow is hypocellular, resembling AA, while glycophosphatidylinositol-deficient PNH cells can be detected in up to 5% of MDS and in >50% of AA patients. Results of recent studies offer a resolution to this overlap: MDS pathogenesis commonly has an autoimmune component and clonal haematopoiesis can be demonstrated in most cases of AA. Two explanations have arisen to explain the association of PNH with these disorders. It is hypothesised that PNH haematopoietic stem cells are relatively resistant to T cell attack and therefore have a competitive advantage in this context. Alternatively, it has been demonstrated that mutations associated with MDS are commonly present in PNH stem cells; such mutations could provide the PNH clone with an autonomous growth advantage. The authors suggest that these mechanisms may be necessary for the development of PNH in all cases, even when PNH occurs in the absence of MDS or AA. Finally, identification of a PNH clone is a predictive and prognostic biomarker in AA and MDS, adding important information for treatment and follow-up.
INTRODUCTION
Bone marrow failure (BMF) occurs when the production of blood cells of one or more lineages by the bone marrow (BM) diminishes to such an extent that cytopenia ensues. Severe BMF is life- threatening, owing to the risks of infection and haemorrhage. Failure of haematopoiesis can result from a variety of causes. In the paediatric age group, 30% of cases of BMF are inherited syndromes (Shwachman–Diamond syndrome, Fanconi anaemia, the telomeropathies, and others). Exposure to ionising radiation, a wide variety of drugs, and certain toxins (e.g., benzene) and systemic illnesses, such as infection and multisystem autoimmune disorders, can all result in transient or permanent BMF. In this review the authors consider three acquired BMF syndromes: aplastic anaemia, myelodysplastic syndrome, and paroxysmal nocturnal haemoglobinuria (PNH). These syndromes appear, on the surface, to be distinct disorders, but have been revealed to be closely interconnected.
PAROXYSMAL NOCTURNAL HAEMOGLOBINURIA, APLASTIC ANAEMIA, AND MYELODYSPLASTIC SYNDROME: DISORDERS OF BONE MARROW FAILURE WITH DISTINCT PATHOBIOLOGY
Paroxysmal Nocturnal Haemoglobinuria
Approximately 1–2 people per million per year are diagnosed with PNH, a rare clonal disorder of haematopoietic stem cells (HSC). PNH is characterised by intravascular haemolysis, pancytopenia, and thrombosis, and, although it is considered a benign disease, can be life-threatening.1 In PNH, somatic mutation of PIGA occurs in a single HSC, resulting in failure of biosynthesis of the glycophosphatidylinositol (GPI) anchor. This structure serves as the attachment point for diverse cell-surface proteins, including the complement defence proteins CD55 and CD59.
This detailed understanding of the biology of PNH has enabled the development of highly reliable flow cytometry assays for the detection of PNH cells in peripheral blood.2 A simple two-colour assay using carefully selected monoclonal antibodies recognising CD235a and CD59 was developed to detect PNH red blood cells (RBC) at a sensitivity of 0.002%. PNH leukocytes are detected using specific monoclonal antibodies to gate monocytes or granulocytes combined with monoclonal antibodies that detect GPI-linked leukocyte antigens (e.g., CD157), along with FLAER, a reagent that binds directly to the GPI anchor. These assays have a lower limit of quantification of 0.02–0.05% for neutrophils, and a somewhat lower sensitivity for monocytes. The routine availability of these assays has revolutionised the diagnosis of PNH and has been crucial in uncovering the relationships of PNH with AA and MDS.
Expansion of the PIGA-mutant HSC clone leads to production of blood cells that lack CD55 and CD59 and are, therefore, very sensitive to lysis by complement proteins, resulting in intravascular destruction of RBC and depletion of nitric oxide, because this compound is scavenged by free haemoglobin. Reduction of the plasma nitric oxide concentration, combined with direct interactions between the complement and coagulation pathways, lead to the clinical complications of PNH: fatigue, dysphagia, abdominal pain, pulmonary hypertension, kidney injury, and a virulent hypercoagulable state. Although the clonal dynamics of PNH are not clearly understood, two hypotheses exist as to how PNH HSC clones survive and expand within the BM: relative growth advantage (intrinsic) and immune escape (extrinsic) hypotheses; these will be highlighted later.
Aplastic Anaemia
AA is a rare BM disorder (incidence: 2–3/1,000,000/year) characterised by destruction of haematopoietic stem and progenitor cells, leading to BMF.3 Although some cases of AA are related to exposure to toxins (e.g., benzene) or to rare inherited mutations (e.g., dyskeratosis congenita), the majority of cases are the result of T cell-mediated autoimmune destruction of HSC, leading to uniformly reduced production of early progenitor cells and CD34+.4 Thus, AA patients present with pancytopenia and histologically aplastic (empty) BM. The development of AA is promoted by augmented immune states, such as pregnancy, toxins, and viral infection.5
Myelodysplastic Syndrome
Myelodysplastic syndrome (MDS) is a heterogeneous group of malignant clonal BM disorders characterised by dysplasia of haematopoietic cells in the BM and peripheral blood, and ineffective haematopoiesis, leading to BMF and cytopenias, and progressing to acute myelogenous leukaemia (AML) in 25% of patients. MDS is more common than PNH and AA, with an incidence of 50 persons per million per year, but in individuals over the age of 70 years its incidence rises sharply to 200–400 per million per year.6 The highly variable natural history of the disease presents challenges in diagnosis and effective and timely treatment.6 Although MDS is typically associated with BM hypercellularity, in ˜10% of cases the BM is hypocellular. This variant, known as hypocellular MDS (hypoMDS), is clinically and pathologically similar to AA (with the exception of the presence of dysplasia) and responds similarly to immunosuppressive treatment.
INTER-RELATIONSHIP BETWEEN PAROXYSMAL NOCTURNAL HAEMOGLOBINURIA, APLASTIC ANAEMIA, AND MYELODYSPLASTIC SYNDROME: COMMON ASSOCIATIONS AMONG RARE DISEASES
With the development of reliable high-sensitivity flow cytometric assays, it is now clear that, despite the rarity of PNH, PNH clones can be detected in a surprisingly sizeable proportion of patients with AA and MDS.2 The biology that underpins these associations has not been elucidated fully. However, observations on the clinical significance of the finding of PNH clones in AA and MDS, and emerging data obtained through the application of new genomics techniques, allow us to create a working model of the inter-relationship between these diseases.
Aplastic Anaemia and Paroxysmal Nocturnal Haemoglobinuria
AA is strongly associated with PNH. When highly sensitive flow cytometry protocols are used, PNH cells are detectable in 40–59% of AA patients.7-9 The majority of the PNH clones are small, affecting <1% of granulocytes, and no correlation is seen between the presence or absence of PNH cells and the severity of AA. However, the presence of PNH cells is predictive of both response to immunosuppressive therapy and overall survival.
The behaviour of PNH clones in AA patients who undergo immunosuppressive therapy is worthy of note. In a series of 207 patients with severe AA who were treated with antithymocyte globulin and cyclosporine, 83 were found to have a detectable PNH clone.8 In 30 of these patients, the PNH clone number increased after treatment, and treatment for PNH was required in 7 patients. Because of the risk of development of clinically important PNH, as well as the predictive and prognostic value of the detection of a PNH clone, flow cytometric analysis for the presence of PNH cells is recommended for all patients diagnosed with AA.10
Although AA is an autoimmune disease, it has long been understood that it possesses some clonal characteristics. Clonal cytogenetic abnormalities are observed in 10–15% of AA patients,11 and in 15% of AA cases patients go on to develop the indisputably clonal diseases MDS or AML. Analysis by single nucleotide polymorphism (SNP) array shows loss of heterozygosity in 15% of AA cases, with many of these affecting the HLA locus, and X-chromosome inactivation studies show evidence of clonal haematopoiesis in 38% of female AA patients.11
Recent studies have used modern genomic techniques to detect somatic mutations in haematopoietic cells in AA. Targeted sequencing of genes, combined with SNP array karyotyping and PNH flow cytometry in 150 AA patients, revealed clonal haematopoiesis in 55% of patients,12 while an approach combining whole-exome sequencing, targeted sequencing, and SNP array karyotyping in 439 patients showed clonal haematopoiesis in 47%.4 In addition to mutations of PIGA and loss of heterozygosity in the vicinity of the HLA locus, somatic mutation was prevalent in genes known to be involved in myeloid malignancy: ASXL1, DNMT3A, BCOR, TET2, JAKs, RUNX1, TP53, and MPL. While PIGA and BCOR mutations, which tended to occur together, were associated with good response to immunosuppressive therapy (IST) and favourable prognosis, patients with myeloid mutations had poor response to IST, poor prognosis, and a higher rate of MDS and AML.4,12
Myelodysplastic Syndrome and Paroxysmal Nocturnal Haemoglobinuria
A strong association also exists between PNH and MDS, although the proportion of MDS cases in which a PNH clone is detectable is much smaller (at ˜2-5%); as for AA, PNH clones found in association with MDS tend to be small (<1% in 45% of cases).13 The strength of this association is stronger for the hypocellular variant of MDS. While hypoMDS accounts for 10% of MDS, ˜70% of PNH+ MDS is hypocellular,14 and a PNH clone can be detected in 40% of cases of hypoMDS.15 As for AA, the presence of a PNH clone in MDS has been reported to be predictive of response to IST,16,17 although this association has not been evident in all studies.
DYNAMICS OF PAROXYSMAL NOCTURNAL HAEMOGLOBINURIA IN BONE MARROW FAILURE: A GAME OF CLONES
In order for a PNH to arise, a PIGA-mutated haematopoietic clone must arise, persist, and expand. However, it appears that although PIGA-mutated clones commonly arise, most do not persist, but instead spontaneously extinguish over the following weeks.18 So why do clones occasionally persist and expand to result in classical PNH?
To account for the persistence and expansion of PNH clones, two hypotheses have arisen: the ‘intrinsic growth advantage’ hypothesis and the ‘immune escape’ hypothesis. The plausibility of the intrinsic growth advantage hypothesis diminished significantly when experimental evidence showed that PIGA-mutant HSC did not possess an intrinsic growth advantage over wild-type stem cells in vitro or in vivo. Consequently, the immune escape hypothesis gained favour.19 This hypothesis (Figure 1A) postulates that, in the context of T cell-mediated autoimmune attack on normal HSC, PIGA-mutant HSC gain a competitive advantage because they are less vulnerable to immune attack owing to loss of the GPI and its associated proteins. The precise mechanism of this reduced susceptibility has not been determined, but could be consistent with, for example, a scenario in which the autoantigen target of T cell attack or an important coregulatory molecule that modulates cell-mediated cytotoxicity is a GPI-linked protein. The immune escape hypothesis is exemplified by the highly prevalent emergence in AA of haematopoietic clones with loss of heterozygosity at the HLA locus.20 This type of mechanism would fit well with the observed strong association between PNH and AA, and with hypoMDS.
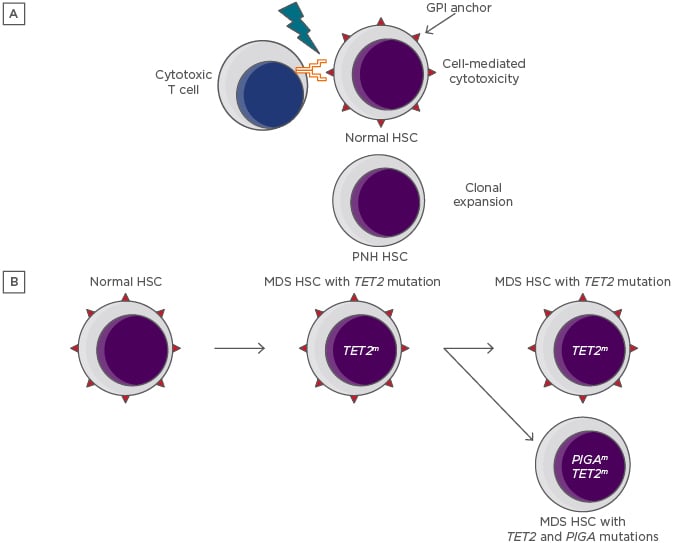
Figure 1: Depictions of the immune escape and intrinsic growth advantage hypotheses for the persistence and expansion of paroxysmal nocturnal haemoglobinuria clones.
A) The immune escape hypothesis. A GPI-dependent T cell-mediated autoimmune attack applies selective pressure on the HSC population. Lacking GPI and its associated proteins, rare PIGA-mutant HSC are relatively resistant to the attack and undergo clonal expansion. B) The intrinsic growth advantage hypothesis. An HSC that already carries a myeloid mutation that confers a growth advantage acquires a PIGA mutation, resulting in the creation of a PNH subclone.
GPI: glycophosphatidylinositol; HSC: haematopoietic stem cells; MDS: myelodysplastic syndrome; PNH: paroxysmal nocturnal haemoglobinuria.
Recent data have led to a resurgence of the intrinsic growth advantage hypothesis. Shen et al.21 discovered unexpected complexity in the genomic landscape of classical PNH by executing whole-exome sequencing and targeted deep sequencing of 61 genes that are commonly mutated in myeloid malignancies.21 Of 60 patients with a diagnosis of classical PNH, 24 were found to have somatic mutation of genes other than PIGA. These data supported an analysis of the clonal dynamics of PNH wherein additional mutations are present, of which three categories were defined. In the majority of cases, a PIGA mutation arose as a secondary event, creating a PNH subclone of an ancestral HSC clone. In other cases, the PIGA mutation was the founding event, and acquisition of myeloid mutations led to clonal evolution, or the PIGA and myeloid-mutant clones were independent of one another. These observations bolster the intrinsic growth advantage hypothesis by pointing to the possibility that PNH clones could gain a growth advantage by hitchhiking with certain myeloid mutations (Figure 1B).
The immune escape hypothesis is consistent with the coexistence of PNH clonal haematopoiesis in AA but is more challenging to reconcile with the expansion of PNH clone size that is frequently seen following treatment of AA with IST.22 After IST, the BM is repopulated from a small number of HSC. In this situation, an HSC clone that possesses an intrinsic growth advantage or a contingent advantage related to immune escape will tend to expand. Immunosuppression would be expected to reverse the environment condition, such as an immune attack, that is responsible for the selective advantage enjoyed in this model by PIGA-mutant HSC.
New data may allow this apparent paradox to be explained without invoking a role for acquired mutations.23 In this study, PNH clone behaviour was studied in 319 patients with AA who received IST. Clone expansion was seen only in patients who had detectable PNH cells prior to IST and was less frequent among patients treated with ATG than those treated with less intensive immune suppression. This supports a model in which successful and complete immunosuppression results in the disappearance of AA-associated PNH clones, but partially effective treatment supports their outgrowth. It should be noted that these data are consistent with the expansion of a residual HSC possessing both a PIGA mutation and a myeloid mutation, such as the common TET2 mutation, which confers a clonal advantage. The authors would expect this mode of clonal selection to be comparatively infrequent.
MDS is a disease characterised by clonal dominance but also, particularly in its hypocellular variant, is in part immune-mediated.24 The persistence of small PNH clones in MDS may largely be sustained by immune escape, a notion supported by the observation that the presence of a PNH clone predicts response to IST. However, the authors conjecture that the intrinsic growth advantage model may also commonly apply: an MDS clone that already possesses an intrinsic growth advantage acquires a PIGA mutation, resulting in a persistent PNH subclone.
These thoughts may also be applied to classical PNH that arises in the absence of any clinically apparent BMF (Figure 2). Here, the PNH clone must, in order to persist and expand, also gain a competitive advantage over normal HSC. The authors propose that in such cases, which account for most cases of PNH, there is always either a concurrent subclinical autoimmune suppression of normal haematopoiesis or clonal haematopoiesis of indeterminate potential/age-related clonal haematopoiesis. In the former scenario, the PNH clone expands via immune escape and in the latter by intrinsic growth advantage.
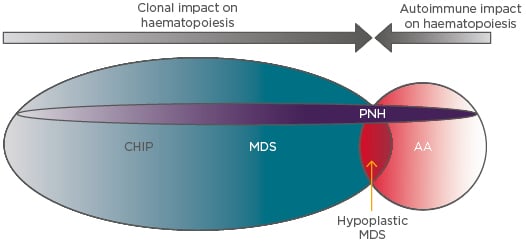
Figure 2: The interplay between the intrinsic and extrinsic influences on paroxysmal nocturnal haemoglobinuria clonal expansion.
In AA the dominant influence is T cell-mediated autoimmune attack, which permits the outgrowth of relatively resistant GPI-negative HSC. This mechanism may also apply in MDS, particularly in the hypocellular variant, which is clinically and pathologically very similar to AA. In MDS the presence of myeloid mutations that confer clonal advantage (e.g., by enhancing HSC self-renewal) also allow concurrent PNH mutations to be fixed and expand in the HSC population. Clonal haematopoiesis of indeterminate potential or subclinical autoimmune HSC attack may also provide support through the same mechanisms for expansion of PNH clones, leading to classical PNH, in which PNH clones expand in the absence of clinicopathological evidence of AA or MDS.
AA: Aplastic anaemia; CHIP: clonal haematopoiesis of indeterminate potential; GPI: glycophosphatidylinositol; HSC: haematopoietic stem cells; MDS: myelodysplastic syndrome; PNH: paroxysmal nocturnal haemoglobinuria.
If it is the case, as the authors have suggested, that the presence of an intrinsic or extrinsic competitive advantage over normal HSC is a necessary condition for the persistence and expansion of PNH clones, then the application of advanced diagnostic techniques might support refined or novel management approaches in classical PNH. For example, patients who are shown by genomic analysis to have a PNH clone that also carry myeloid mutations would be followed more closely for the development of MDS or AML and may be advanced as candidates for curative treatment with allogeneic stem cell transplantation. Conversely, for patients with classical PNH in whom no myeloid mutations were found, it might be deduced that an underlying T cell-mediated autoimmune attack on HSC had supported the expansion of the PNH clone. In such patients, it may be predicted that aggressive IST could lead to the eradication of the condition supporting the persistence and expansion of the PNH clone by removing it, thus potentially curing the disease and removing the need for lifelong anticomplement therapy.
CLINICAL IMPLICATIONS OF PAROXYSMAL NOCTURNAL HAEMOGLOBINURIA CLONES IN MYELODYSPLASTIC SYNDROME AND APLASTIC ANAEMIA
The detection of a PNH clone in AA or MDS patients is of established clinical value, since the presence of even minor populations of PNH cells in patients with AA or MDS is an important predictor of a higher rate of response to IST and superior overall survival.7,13,14,16,25 This is important in lower-risk MDS patients, in whom active therapeutic options are limited. In addition, PNH clones in AA may expand and cause life-threatening PNH. Therefore, identification and surveillance of PNH clones is recommended in all patients diagnosed with AA.10,14 These principles are illustrated in the following brief clinical scenarios.
Scenario 1
A 33-year-old man, a recent immigrant with a history of severe aplastic anaemia diagnosed 8 years ago and successfully treated in his home country with IST, now presents with dark urine, dyspnoea, and severe anaemia. Initial investigations reveal pulmonary embolism and iliac vein thrombosis. His lactate dehydrogenase is elevated 9-times the upper limit of the normal range. Flow cytometry for PNH reveals GPI-deficient white blood cells (WBC) (FLAER-/CD157-): CD15+ neutrophils: 54.7%; CD64+ monocytes: 76.3%. Total GPI-deficient RBC (CD235a+/CD59-): 22.5% (Type III RBC: 14.4%; Type II RBC: 8.1%).
This case illustrates the importance of testing for the presence of PNH cells in all patients diagnosed with aplastic anaemia. In a significant proportion of patients who initially have a small PNH population, the size of the PNH clone expands after IST and may become clinically significant. In this patient, development of PNH led to a catastrophic clinical presentation that could easily have been fatal; this situation may have been avoided by surveillance, timely recognition of the expanding PNH clone, and implementation of anticomplement therapy.
Scenario 2
A 50-year-old man who is otherwise healthy and takes no medication presents with pancytopenia. Investigations, including a BM examination, lead to a diagnosis of severe aplastic anaemia. Flow cytometry for PNH reveals the presence of a small clone GPI-deficient WBC (FLAER-/CD157-): CD15+ neutrophils: 1.3%; CD64+ monocytes: 1.5%. Total GPI-deficient RBC (CD235a+/CD59-): 0.01% (Type III RBC: <0.01%; Type II RBC: <0.01%).
The best first-line therapy for this patient may be debated. Current recommendations are for allogeneic stem cell transplantation in patients <35 years old and IST for patients >50 years old, while either approach is acceptable for patients 35–50 years.10 This patient has no comorbidities but is at the upper limit for his age range for stem cell transplantation and so is at higher risk for transplant-related morbidity and mortality. However, the detection of a PNH clone in this case indicates that this patient has a higher-than-average chance of responding to IST, and a better-than-average prognosis; these facts point toward choosing IST as first-line therapy. If he is treated with IST, this patient will require regular surveillance of the size of his PNH clone.
Scenario 3
A 57-year-old woman presents with pancytopenia (red cell transfusion dependence, platelets <20×109/L and neutrophils <0.5 x109/L). BM examination reveals multilineage dysplasia with markedly decreased BM cellularity (10%) and normal karyotype. The International Scoring System risk category is intermediate-1, and the Revised International Scoring System risk category is intermediate. Flow cytometry testing for PNH shows GPI-deficient WBC (FLAER-/CD157-): CD15+ neutrophils: 2.5%, CD64+ monocytes: 2.8%. Total GPI-deficient RBC (CD235a+/CD59-): 1.04% (Type III RBC: 1.03%; Type II RBC: 0.01%).
Patients with lower risk MDS who experience multiple cytopenias present a difficult therapeutic problem. Medical therapies for this condition are limited; some, like recombinant human erythropoietin and lenalidomide, are effective only in treating anaemia, while others, such as hypomethylating agents, are significantly inconvenient for patients who are expected to survive several years, since they require indefinite cycles of treatment. Allogeneic stem cell transplantation may be an option for some patients, although a decision analysis suggests that this modality of therapy results in a net loss of years of life.26 The detection of a small PNH population in this patient suggests IST as an alternative approach. In this clinical situation, IST with ATG and cyclosporine can yield a durable trilineage response with acceptable toxicity.
CONCLUSION
Traditionally considered to be distinct BMF disorders with distinctive pathophysiology, current evidence suggests that PNH, AA, and MDS feature a complex interplay between clonal haematopoiesis and T cell-mediated autoimmune attack on normal HSC. The clonal development of PNH, both in the context of AA, MDS, or classical PNH, is currently explained by two hypotheses: the relative growth advantage (intrinsic) hypothesis and the immune escape (extrinsic) hypothesis. Evidence for each of these hypotheses comes from clinical observations and new data from next-generation sequencing analysis. These data have implications for the clinical management of AA and MDS, as well as for understanding the biology and potentially guiding the treatment of classical PNH. Future research will be required to test the contention that either co-operating mutations or autoimmune attack must always be present for PNH to develop and if in some cases PNH can be successfully eradicated by intensive IST.





