Editorial Note: This article was temporarily removed from 27.11.24 to 14.08.25 for review. The review has been completed, and the content remains unchanged from its original publication.
![]()
Meeting Summary
At the 2024 European Association of Neurology (EAN) Congress, one satellite symposium discussed the recognition, diagnosis, and treatment of Friedreich ataxia (FA), the most common hereditary ataxia. This condition is characterised by progressive neurodegeneration, multisystem complications, loss of ambulation, and reductions in the ability to carry out activities of daily living (ADL). For many, there is also a premature death. FA is caused by guanine-adenine-adenine triplet (GAA) repeat expansions in the gene FXN. This codes for the protein frataxin, loss of which is associated with impaired mitochondrial function, increased sensitivity to oxidative stress and reactive oxygen species levels, increased inflammation, and cell death.
Decreased frataxin leads to the symptoms of FA, including increasing spasticity, pain, dysphagia, cardiac problems, speech impairment, pes cavus, and scoliosis. The speakers highlighted how delays in diagnosis can occur when FA is mistaken for other ataxias and they called for the use of genetic and biochemical testing early in the patient pathway. This is best accomplished by prompt referral to specialists in ataxia. Treatment and care for patients with FA, along with their families, require a multidisciplinary approach involving allied healthcare professionals, among other specialists. Effective communication and support amongst such networks is key to providing individualised treatment where a patient’s health and disease progression are regularly monitored and associated conditions are treated appropriately. Currently, the only approved FA-specific drug treatment in the EU and the USA for patients 16 years and older is omaveloxolone. Clinical trials of this drug have shown that it can provide a sustained benefit in slowing disease progression over a 3-year period in patients aged 16 years and older. This benefit is particularly evident when omaveloxolone prescription is not delayed. In the future, other pipeline drugs are expected to add to potential disease-slowing treatments for FA.
![]()
Introduction
FA is a complex, autosomal recessive, cerebellar ataxia involving progressive degeneration of central and peripheral nervous systems, including loss of several types of neurons and associated nerves. This presents as effects in cardiac, musculoskeletal, and endocrine systems.1,2 While FA is considered rare, it is the most common hereditary ataxia, accounting for around 50% of all ataxia cases.1 In Caucasian individuals, overall prevalence is generally cited as being 1 in 20,000–50,000.3 However, studies from individual countries indicate that prevalence varies from 0.1 in 100,000 in Finland to 4.7 in 100,000 in Northern Spain, with figures for other European countries falling between these two.4-6
As part of a satellite symposium at EAN 2024, two experts in FA, Mathieu Anheim and Jörg Schulz, discussed the recognition and diagnosis of FA, along with current treatments. The symposium started with video testimonies from people involved in FA. Bart Jan, the father of a 20-year-old with FA, discussed how the biggest challenge was getting a diagnosis and how, for them, “it took us close to 6 years, travelling from doctor to doctor, appointment to appointment”. Stephan Rouillon, from the French Association for Friedreich Ataxia,7 discussed the importance of patients “finding the right specialists and understanding what can be done to improve their life.” Susan Milman, from Euro-Ataxia,8 emphasised how patients and families “need information about how to live with the condition and how not to let the condition destroy their lives.”
Recognising the Signs and Symptoms of Friedreich Ataxia
Many key clinical features of FA are classed as neurological.1,2,9,10 Of note, said Anheim, is combined proprioceptive and cerebellar ataxia, the latter of which is associated with slurred speech due to cerebellar dysarthria. There is also the abolition of tendon reflexes and hypopallesthesia due to sensory neuronopathy or ganglionopathy, which can be identified with electroneuromyography. Additionally evident may be bilateral extensor plantar reflexes, flexor spasms, urinary urgency/incontinence, and square wave jerks. In the context of mitochondrial disease, Anheim discussed extra-neurological signs of FA. These include hypertrophic cardiomyopathy, scoliosis, pes cavus, diabetes, optic neuropathy, and deafness.1,2,9,10 Many of the symptoms associated with these features are depicted in Figure 1.
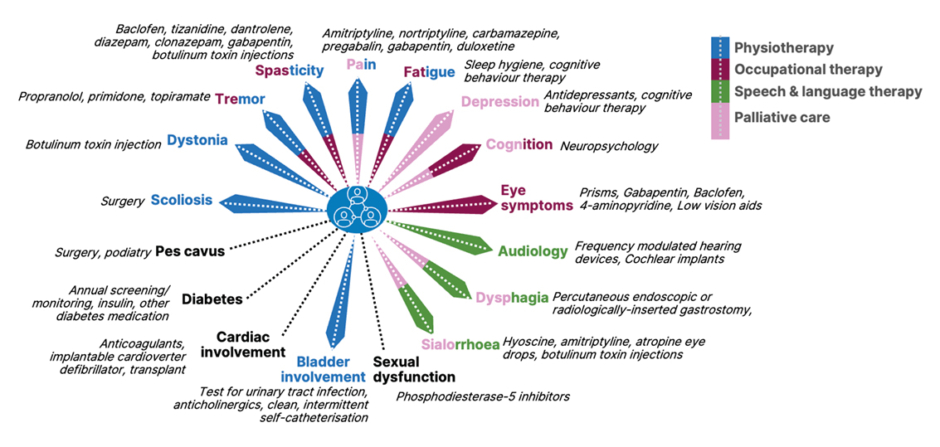
Figure 1: Friedreich ataxia symptoms and their management.9-12
Partially adapted from de Silva et al.,11 2019
Progression of Friedreich Ataxia
The onset of FA symptoms is typically in childhood, aged 8–14 years (‘typical-onset FA’), though in some it may be earlier (0–7 years, ‘early-onset FA’), or more into adolescence/young adulthood (15–24 years), or even later (>24 years, ‘late-onset FA’). In general, the progression of ambulatory factors of FA is faster in early-onset FA compared to any other groups.13 While cardiomyopathy and diabetes may occur early in patients with early- and typical-onset FA, these may be at a lower rate or absent in patients with late-onset FA. Additionally, foot deformities due to pes cavus, other cardiovascular issues, and pronounced scoliosis also occur at a much lower rate in patients with late-onset FA.9
Over the years, initial unsteady gait and general clumsiness gradually become difficulty in standing and walking as well as problems with falls. The frequency and intensity of these symptoms increase over time, leading to the need for ambulatory walking aids, and at later stages, seated mobility aids.9,14
Ambulation loss, which is seen as a critical milestone in FA progression,15 usually occurs by the mid-to late-20s in patients with early-onset FA.10 However, FA burden is not just in ambulation, but through functional disabilities. For example, curtailment of the ability to carry out ADLs such as feeding oneself or drinking; marked cerebellar dysarthria leading to difficulties in speaking clearly and being understood; and sight fixation problems due to square wave jerks, among others.6,10,16 In patients with late-onset FA, there is usually a longer disease duration before ADLs are impacted and seated mobility aids are required.17
Overall, prognosis is poor, and the mean age of death is 36.5 (range: 12–87) years.1,18 Primarily, the cause of death in FA is due to cardiac dysfunction, predominantly in the form of arrhythmia or congestive heart failure.18
Genetics of Friedreich Ataxia
In FA, GAA repeat expansions in the gene FXN on chromosome 9 are inherited in an autosomal recessive pattern. Typically, there are between 200 to over 900 GAA units in patients with FA, compared to 7–22 units in non-affected controls. GAA repeat expansion impairs normal expression of the FXN gene, which codes for frataxin, a protein involved in mitochondrial iron homeostasis and biosynthesis of iron-sulphur clusters, impacting their assembly and transfer to mitochondrial components.19,20 Most patients with FA are homozygous for GAA expansion (96%), with a minority being compound heterozygous, where there is GAA expansion on one allele and a point mutation or exon deletion on the other (4%).21,22
Decreased viable FXN expression leads to around 70−95% reduction in frataxin production compared to levels in unaffected people.23 Decreased frataxin levels result in impaired mitochondrial function, increased sensitivity to oxidative stress, and increased levels of reactive oxygen species. This can lead to increased inflammation, and ultimately, cell death.19,21,24
In general, lower frataxin levels correlate with greater disease severity.19,21 In homozygous patients, the amount of frataxin manufactured generally corresponds to the number of GAA repeats on the least-affected allele. That is, there is an inverse correlation between the length of the allele with the shorter GAA expansion and frataxin levels, as well as with age of onset, and disease progression rate. This means that earlier disease onset and faster progression are associated with longer alleles, and those with shorter GAA expansion on this allele typically have later disease onset, sometimes into middle or even older age.17,19,21-25
Diagnosis of Friedreich Ataxia
Genetic and Biochemical Testing
Genetic testing should be carried out in all patients displaying any signs of cerebellar ataxia, said Anheim. He discussed how, in his experience, GAA expansion must be searched for with specific molecular analysis as it cannot be found using targeted gene panel or whole exome sequencing. Genetic testing is vital to distinguish FA from other ataxias with sensory neuropathy that may mimic FA. These include abetalipoproteinaemia, due to mutations in the MTP (microsomal triglyceride transfer protein) gene, and ‘ataxia with vitamin E deficiency,’ due to mutations in the alpha-TTP (tocopherol transfer protein) gene. Genetic testing can also distinguish FA from ataxias without peripheral neuropathy, such as Neimann-Pick disease Type C (NPC), due to mutations in the NPC1/2 gene, and from ataxias with sensory and motor neuropathy, such as ataxia telangiectasia (AT), due to mutations in the ATM (AT mutated) gene.26
Anheim also discussed how assessing biomarkers may be useful for distinguishing FA from treatable diseases with symptoms resembling FA. For example, vitamin E is markedly reduced in ‘ataxia with vitamin E deficiency’,27 and is malabsorbed, along with fats and other fat-soluble vitamins, in patients with abetalipoproteinaemia.28 Further, NPC can be distinguished by raised plasma oxysterol levels,29 and ‘ataxia with oculomotor apraxia Type 2’ can be distinguished by raised serum alpha-fetoprotein levels.30 Brain scan investigation in patients with FA may reveal upper cervical cord atrophy but little-to-no cerebellar atrophy.11
Clinical Symptoms
Using case studies, Anheim pointed out examples of what can be deduced in a clinical examination. For example, sway should be detected when a patient stands with their feet together (which worsens when their eyes are closed), and hypermetria (overshooting the intended goal) should occur when a patient tries to move their heel down their shin or touch their nose then, sequentially, the examiner’s fingers. Another example was of a patient having difficulties with balance and gait when turning around during a walking test. They also displayed saccadic pursuits on ocular examination, and disappearance of ankle, knee, and extensor plantar reflexes.
Such symptoms can be assessed and tracked using validated neurological rating scales. The modified FA Rating Scale (mFARS) helps assess neurological deficits in four domains: bulbar function, upper limb coordination, lower limb coordination, and upright stability. Combined scores range from 0−93 (best to worst); more details are shown in Figure 2.31,32 In another case presented by Anheim, a patient with an mFARS score of 58 had lost most of their ambulation ability and was a wheelchair user. On standing, they could only walk with the support of a person and a mobility aid, and they had difficulty coordinating their lower limb movements.
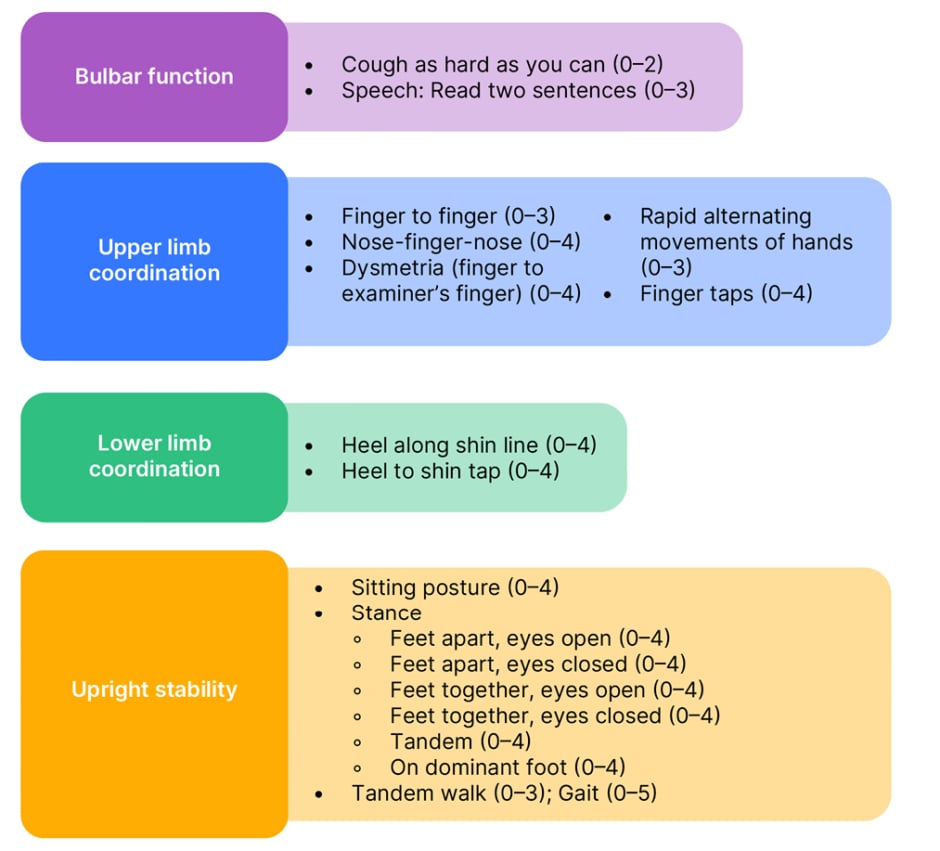
Figure 2: Modified Friedreich’s ataxia rating scale (mFARS) (scores from best [0] to worst in 0.5 increments).31,32
Adapted from Rummey C et al.31 and Friedreich’s Ataxia Research Alliance32
A slightly faster but less FA-specific scoring method is the Scale for the Assessment and Rating of Ataxia (SARA). This can be used to assess stance, gait, limb kinetic function, sitting, and speech. Combined scores range from 0–40 (best to worst).33
Avoiding Diagnostic Delay
As stressed though testimony in the introduction to this symposium, it is important to recognise that many patients and caregivers with FA wait a long time for a diagnosis with the journey including visits to primary then a number of specialist care providers, mis- and missed diagnoses, genetic testing, and involvement of patient organisations along the way.25,34,35 Accordingly, Anheim emphasised that a crucial first step to diagnosis is to swiftly refer the patient to a specialist neurologist/neuropaediatrician who understands how to diagnose FA and can help put together the best care package for the individual. As prognosis may be poor, and especially for those with early-onset FA, progression can be reasonably rapid, it is vital that patients are diagnosed and have a care plan in place as soon as possible so they can live a more supported and comfortable life.1,16,36
Treatment
FA is a multisystem disorder that needs a multisystem approach with regard to clinical management and care.1,11,16 With this in mind, in 2022, best practice recommendations were developed in collaboration with healthcare professionals, researchers, and patients with FA and their families/caregivers.12 As illustrated in Figure 1, overall, there is a need for physiotherapy, occupational therapy, speech and language therapy, and later in life, palliative care. Treatment should be individualised according to symptoms and needs. For example, surgery may be needed for scoliosis and pes cavus, and muscle relaxants, pain relief, and botulinum toxin injections may be needed for dystonia, tremors, spasticity, pain, eye symptoms, and sialorrhoea. Further, cognitive behaviour therapy may be of use for symptoms such as fatigue and depression, and antidepressants may be prescribed for the latter, as well as, in some cases, for pain.11
Schulz discussed how individualised treatment may be coordinated by a primary care physician or a neurologist, and how allied specialist teams are necessary to help a patient and their family over the course of FA.11,37 A care review should be offered every 6−12 months, wherein a general or specialist ataxia neurologist monitors disease progression and manages comorbidities as needed. The multidisciplinary team can also provide individualised follow-ups, for example in the realms of diabetes care, cardiomyopathy, scoliosis, and other treatable or manageable symptoms. Vital for the patient and their family is effective, mutually supportive communication among the healthcare professionals providing care.11,37
Omaveloxolone
As discussed previously, deficits in frataxin in FA lead to impaired mitochondrial function and increased oxidative stress and inflammation.19,24 Also seen in the cells of people with FA, and in pre-clinical models, is suppression of levels and activity of the protein nuclear factor erythroid 2-related factor (Nrf2). This is important in patients with FA as impaired Nrf2 signalling correlates with low levels of frataxin expression. Nrf2 can also induce the expression of a number of cytoprotective proteins involved in supporting mitochondrial function, bioenergetics, and proteostasis, and in inhibiting inflammatory responses and curtailing oxidative stress.38
Usually, Nrf2 levels are kept low due to degradation coordinated by kelch-like ECH-associated protein 1 (Keap1).38 The drug omaveloxolone, a semi-synthetic derivative of oleanolic acid, has been developed to help counter such actions by increasing the availability of Nrf2. Omaveloxolone can bind to Keap1 and inhibit its ability to target Nrf2 for degradation, allowing Nrf2 to translocate into the nucleus and regulate the transcription of its target gene. Omaveloxolone can also inhibit inflammation directly by inhibiting proinflammatory signalling pathways and reducing the production of proinflammatory mediators,39 and both in vitro and in vivo, it has been shown to restore mitochondrial activity and decrease oxidative stress.40
Omaveloxolone is currently the only drug licenced for the treatment of FA in the EU and USA. It is indicated for patients aged 16 years or older at a dose of 150 mg/day.41 The ’MOXIe’ clinical trial programme for evaluating omaveloxolone in patients aged 16−40 years included a two-part, Phase II, double-blind, randomised, placebo-controlled trial.42 Part 1 was a dose-ranging study, over 12 weeks, assessing the pharmacodynamics, pharmacokinetics, and safety of omaveloxolone doses from 2.5–300 mg/day versus placebo (n=69).43 Part 2, over 48 weeks, assessed efficacy and safety of 150 mg/day omaveloxolone versus placebo.44
In Part 2, the full analysis set used for primary analysis of efficacy included all patients who had at least one postbaseline measurement but did not include patients with pes cavus due to Part 1 results showing outliers in this group.43 At baseline, most participants were ambulatory with mean ± standard deviation (SD) mFARS scores of 40.9±10.4 for the omaveloxolone group (n=40) and 38.8±11.0 for the placebo group (n=42). In this study, a version of the mFARS was used that included two additional assessments of bulbar function taken from the previous, slightly longer, FARS neurological assessment;31 as such scores ranged from 0−99. Section GAA1 repeat length was slightly higher in the omaveloxolone group (mean ± standard deviation, 739.2±214.9) compared with the placebo group (693.8±277.2).44
Results showed that by Week 48, placebo group participant scores worsened (mean ±standard error of the mean; 0.85±0.64; 95% CI: -0.43, 2.13) while those in the omaveloxolone group improved (-1.55±0.69; 95% CI: −-2.93, −0.18). The difference between them (-2.40±0.96; 95% CI: -4.31, -0.50) was significant (p=0.014).44
Of the several components of mFARS, omaveloxolone had the greatest effect on upright limb coordination, followed by upright stability scores.44,45 Such differences are relevant as Shulz discussed how a one-point worsening on scores of upright stability can mean a patient going from having mild ataxia, with little need for safety support, to having definite ataxia, with intermittent need for safety support due to impaired ambulation and the risk of falls. Improvements compared to placebo were also shown in bulbar scores.44 A one-point change here can mean the difference between a patient being able to speak at a mostly understandable level, to one where clear verbal communication is difficult (Figure 3).32
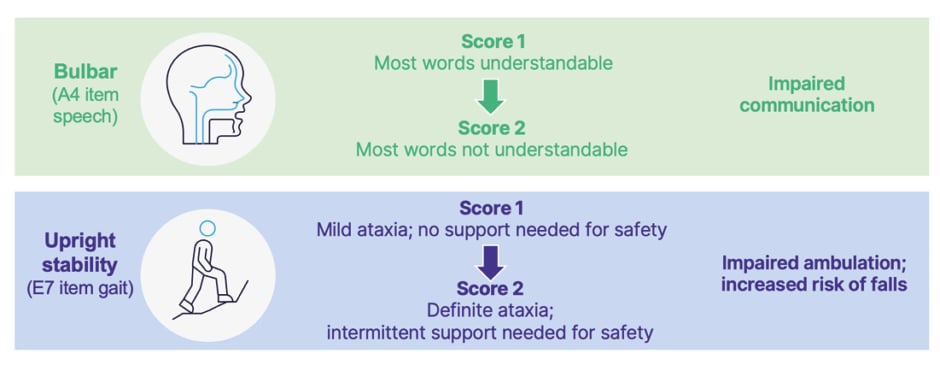
Figure 3: Clinical implications of changes in modified Friedreich’s ataxia rating scale (mFARS) scores.32
Adapted from Rummey C et al.31 and Friedreich’s Ataxia Research Alliance32 A4: Subscale A, instruction 4 on the mFARS, assessing speech; E7: Subscale E, instruction 7 on the mFARS, assessing gait.
Secondary endpoints were analysed using a fixed-sequence hierarchical approach to maintain the familywise overall Type I error rate of 0.05. Using this strategy, numeric, though not statistically significant, improvements were shown in scores of Clinician and Patient Global Impression of Change (PGIC) measures, fall frequency, dexterity, and walking.44 The ‘FA-validated ADL’ (FA-ADL)46 scale showed that omaveloxolone improved scores from baseline and reached nominal statistical significance compared to placebo at Week 48 (p=0.04). Post-hoc analysis of the proportion of patients that improved or worsened in primary and secondary measures at Week 48 showed that a greater proportion of patients in the omaveloxolone group improved, and fewer worsened, on the mFARS, ADL scale, and PGIC measure.44
Overall, omaveloxolone was generally well-tolerated with a serious adverse event (AE) in 6% of the placebo and 10% of the omaveloxolone group. AEs occurring in ≥25% of patients in placebo/omaveloxolone groups, respectively, included headache (25%/37%), alanine aminotransferase (ALT) increase (2%/37%), contusion (37%/33%), nausea (14%/33%), upper respiratory tract infection (29%/28%), and excoriation (23%/26%). Schulz reported that, in general, excess AEs in the omaveloxolone group were limited to the first 12 weeks of treatment, with lower frequencies reported between 12–48 weeks. Discontinuations due to an AE were 4% in the placebo group and 8% in the omaveloxolone group.39,42,44,47,48
In patients treated with omaveloxolone in the MOXIe study, increases above the upper limit of normal (ULN) were observed for ALT or aspartate aminotransferase (AST) (29.4% ≥3x ULN; 15.7% ≥5x ULN) and BNP (13.7% >ULN), with hypercholesterolaemia observed at a rate of 2%. Maximal increase occurred within the first 12 weeks of treatment.41,44,48 This is not unexpected because, as an Nrf2 activator, omaveloxolone has been reported to increase aminotransferase expression, partially due to its pharmacodynamic effects.49
According to prescribing guidelines, ALT, AST, and bilirubin levels should be ascertained prior to omaveloxolone initiation, then monthly for the first 3 months, then periodically as clinically indicated. Omaveloxolone should be discontinued immediately if ALT or AST increases to >5x ULN or to >3x ULN if accompanied by bilirubin >2x ULN. Liver tests should then be repeated. While for patients with severe hepatic impairment (Child-Pugh Class C), use of omaveloxolone should be avoided, in patients with moderate impairment (Child-Pugh Class B), the dose should be 100 mg/day with close monitoring for adverse reactions, with consideration of lowering to 50 mg/day if adverse reactions occur. Increases in low-density lipoprotein and decreases in high-density lipoprotein, as well as decreases in body weight, may also occur and should be monitored and treated according to standard clinical guidelines.41
Open-Label Extension, Follow-on, and Real-World Studies of Omaveloxolone
An ongoing open-label extension (OLE) study is being carried out to assess the long-term safety and tolerability of 150 mg/day omaveloxolone in qualified patients with FA following the completion of Part 1 or Part 2. Participants who were initially in the placebo group in Part 2 were switched to omaveloxolone during the OLE trial.50,51
Results from the OLE include that after 72 weeks, the notable difference between omaveloxolone and placebo in mFARS change from baseline scores shown in Part 1 and 2 remained, favouring the omaveloxolone-omaveloxolone group (n=40) over the placebo-omaveloxolone group (n=42) (least squares mean difference: -2.91, standard error: ±1.44). While by the end of OLE study mFARS scores had increased above baseline in both groups, it was of note that scores did not rise above extension baseline in the omaveloxolone-omaveloxolone group until Week 120 testing. This, said Schulz, indicated that omaveloxolone had a slowing effect on disease progression. Also discussed by Schulz was that, at the end of Part 2, participants were required to enter a washout period of 4 weeks. During this time, mFARS scores in the omaveloxolone-treated group returned to baseline,51 showing that gains in Part 2 had been due to this drug.
Data from the MOXIe trial was also compared to data from the large, global, multicentre, prospective natural history, longitudinal ‘FA Clinical Outcome Measures Study’ (FA-COMS).52 Omaveloxolone was associated with slower disease progression (55% reduction in mFARS) in treated patients compared to the propensity-matched FA-COMS cohort over a 3-year period. In the primary pooled population (136 patients in each group), by Year 3, patients in the FA-COMS matched set progressed 6.6 points on mFARS, whereas patients treated with omaveloxolone in MOXIe OLE progressed 3.0 points (difference: -3.6; nominal p=0.0001).50
The Future of Friedreich Ataxia Treatment
While omaveloxolone is currently the only therapy approved specifically for FA in the EU and USA, a number of other drugs that may improve mitochondrial function, reduce oxidative stress, or modulate frataxin-controlled metabolic pathways, are in human trials. Schulz noted, however, that such drugs only target the results of frataxin loss in FA. To target frataxin itself, trials are also being carried out for therapies that can act as frataxin replacements, stabilisers, or enhancers; increase FA-related gene expression; or replace or edit FA-related genes.53
Conclusion
As FA is a progressive, often life-shortening, condition, this symposium highlighted how diagnosis should occur as soon as possible after ataxia presents, via referral to a specialist centre for clinical evaluation and genetic analysis.2,11,25,34,35,54 FA is characterised by multisystem complications16 so treatment and care, as well as family/caregiver support, requires a multidisciplinary approach involving allied healthcare professionals and others.11 Effective communication and support amongst this multidisciplinary team is key to providing individualised treatment where patient progress is regularly monitored and associated conditions treated appropriately.37
Currently, the only FA-specific approved drug treatment in the EU and USA is omaveloxolone.41 This has been shown to provide a sustained benefit over a 3-year period in patients aged 16 years and older including slowing progression of FA symptoms. Such results were particularly apparent when omaveloxolone use was not delayed.43,44,47,50,51, In the near future, other pipeline drugs are being investigated to address disease manifestations of FA.52






