Meeting Summary
IgA nephropathy (IgAN) is a common form of glomerular disease, with wide heterogeneity of symptom occurrence and progression. Diagnosis is based on kidney biopsy findings. IgAN initiates in the mucosa with development of galactose-deficient IgA1 (Gd-IgA1) and anti-Gd-IgA1 autoantibodies, leading to deposition of these complexes in glomerular mesangium with resulting fibrosis, inflammation, tubulointerstitial scarring, and glomerular injury. This can lead to chronic kidney disease (CKD), kidney failure, and death. IgAN treatment involves optimised supportive care, including individualised strategies to address symptoms, such as high blood pressure control and cardiovascular risks. Drug treatment includes renin-angiotensin-aldosterone system (RAAS) inhibitors and immunosuppressant therapies. While the latter can successfully lower proteinuria, and have a positive effect on estimated glomerular filtration rate (eGFR), adverse effects can limit treatment duration, and increasing proteinuria and decreasing eGFR can return following treatment discontinuation. New formulations of immunosuppressant therapies include delayed-release budesonide with targeted release in the lower part of the small intestine where Gd-IgA1 production occurs. Although treatment with this drug can reduce proteinuria and sustain eGFR levels, similar to other immunosuppressant therapies, effects seem to be predominantly limited to the active treatment period. Targeting a different mechanism, sparsentan is a dual endothelin A receptor (ETA) and angiotensin II receptor type 1 (AT1) blocker that targets endothelin-1 (ET-1) and angiotensin II, both involved in IgAN progression. Initial Phase III trial results show significant differences, favouring sparsentan, compared with the AT1 blocker irbesartan, on proteinuria, with similar adverse event profiles. These agents, and several other drugs in development, will widen the armamentarium of therapies for people with IgAN, which, when used in combination, can target different aspects of IgAN pathogenesis for a more individualised treatment approach.
Introduction
IgAN, which can lead to CKD and kidney failure, is the most common form of primary glomerulonephritis, predominantly, though not exclusively, occurring in young adults.1,2 Prevalence of IgAN, and associated CKD is higher in people of East Asian ancestry.3 While IgAN progression can be slow in some patients, occurring over 20–25 years, in many, progression can be rapid. This means that early diagnosis and treatment is key, as even in patients where development is slow, IgAN occurrence in a young adult can mean progression in their middle age.2,4 Symptoms are heterogeneous, and can range widely from asymptomatic microscopic haematuria to sustained proteinuria and rapid renal function decline.1,2
Diagnosis of IgAN necessitates a kidney biopsy to ascertain the ‘MEST-C’ score, based on a combination of five histological components: mesangial (M) and endocapillary (E) hypercellularity, segmental sclerosis (S), interstitial fibrosis/tubular atrophy (T), and crescents (C).5 Results from MEST-C assessment can be incorporated into prognosis predictions, but, according to Kidney Disease: Improving Global Outcomes (KDIGO) guidelines, “there is insufficient evidence to support the use of the MEST-C score in determining whether immunosuppression should be commenced in IgAN.”4
At the Nephro Update Europe 2023 Congress, An De Vriese, Department of Internal Medicine, Division of Nephrology and Infectious Diseases, AZ Sint-Jan, Brugge, Belgium; and Vladimir Tesař, Department of Nephrology, Charles University, Prague, Czechia, discussed IgAN pathogenesis, current treatments, and how, with the development of several new treatments, IgAN therapy should be individualised according to each patient’s needs.
Pathogenesis of IgA Nephropathy
De Vriese explained how the basic pathogenesis of IgAN can be encompassed within the “four-hit hypothesis.”1,2 In this model, the initiating hit is when elevated circulating levels of Gd-IgA1 occur following a mucosal infection due to, often inherited,6 abnormalities in IgA1 production. The second hit, development of autoantibodies, occurs as Gd-IgA1 lacks galactose residues around the hinge region needed to clear IgA1, and this area is exposed as an autoantigen.1,2,7,8 The third hit occurs when Gd-IgA1 and anti-Gd-IgA1 autoantibodies combine to form immune complexes, leading to the fourth hit, deposition of these complexes in glomerular mesangium. This can result in inflammation and fibrosis, due to the stimulation of mesangial cells; activation of the complement pathway; and secretion of chemokines, cytokines, and extracellular matrix proteins, leading to glomerular injury and tubulointerstitial scarring.1,2,7
Current Treatment of IgA Nephropathy
With regard to the four-hit hypothesis, De Vriese noted: “It is important to realise that all four hits must occur for clinically relevant disease to develop. This implies that solely targeting one factor in treatment will not work; instead, you need a comprehensive approach that targets all the relevant factors.” De Vriese also explained how “you need to judge in an individual patient which element of the pathophysiology is determining the clinical effects you see. Is this proteinuria because of chronical scarring and incomplete restoration of the glomerular filtration barrier, or is this proteinuria the consequence of active inflammatory disease? This will determine which drug(s) you will choose.”
Another difficulty when assessing and treating IgAN, De Vriese explained, is that “there’s an extreme heterogeneity in clinical presentation and prognosis of patients, from isolated microscopic haematuria with very low risk of progressive disease that just requires annual follow-up to, on the other end of the spectrum, rapidly progressive glomerulonephritis that has a dire prognosis that should be treated aggressively. The problem,” De Vriese continued, “is that a large majority of patients are somewhere along this spectrum, and it can be difficult to figure out where exactly they are at.”1,2,9 De Vriese explained how “the challenge in the approach to IgAN is not only to assess the risk of progressive disease, but to try to figure out if a patient has active inflammatory disease, and will likely respond to a course of immunosuppressive treatment.”
To aid IgAN management, the KDIGO Glomerular Diseases Work Group provided updated IgAN treatment guidelines in 2021. These state that “the primary focus of management should be optimised supportive care.” Such care includes individualised strategies for the patient that involve, as needed, blood pressure control, cardiovascular risk minimisation, dietary counselling, smoking cessation, weight control, and exercise.4
Medical management for IgAN in KDIGO guidelines is predominantly based on proteinuria levels. If proteinuria is >0.50 g/day, RAAS inhibitors should be prescribed, regardless of presence of hypertension. If proteinuria is >0.75–1.00 g/day, despite at least 90 days of optimised supportive care, a patient is considered to be at high risk of IgAN progression, and a 6-month course of glucocorticoid treatment can be considered following discussion of potential adverse events. However, it is noted in the guidelines that “clinical benefit of glucocorticoids in IgAN is not established and should be given with extreme caution or avoided entirely in patients with a number of conditions.” These conditions include eGFR <30 mL/min/1.73 m2, diabetes, obesity, latent infections, and severe osteoporosis. For patients who remain at high risk of progressive kidney disease despite maximal supportive care, KDIGO guidelines suggest they “should be offered the opportunity to take part in a clinical trial.”4
These recommendations are partly based on the Toronto Glomerulonephritis registry study, showing that proteinuria is one of the most important modifiable indicators of renal survival in patients with IgAN. This study showed that in patients with <1 g/day proteinuria, rate of renal decline was 90% slower than the mean rate, but increased in patients with higher proteinuria levels.10 This means that, according to Tesař, “if we are able to reduce proteinuria, we should be able to modify the course of the disease.” Indeed, the Valiga study, conducted by Tesař and colleagues, showed that for patients receiving RAAS blockade with or without glucocorticoids, those achieving proteinuria of <1 g/day had more favourable outcomes than patients not achieving this level.11 As such, Tesař said, “we should aim for low proteinuria.” Additionally, Tesař continued, “the longer proteinuria remission is, the better the outcome of the patients.” Decreasing this risk may take a number of years, but once achieved and maintained, the long-term outcome, said Tesař, should be favourable.12
However, while prescribing immunosuppressive treatment based on proteinuria level is useful, De Vriese suggested the importance of “incorporating the presence of persistent microscopic haematuria in your decision-making.” Although this is not proposed in KDIGO guidelines, De Vriese also suggested taking the MEST-C score into account, as the results of one study showed that adding MEST-C to baseline assessments, such as proteinuria, can improve prediction of 50% decrease in eGFR or kidney failure.4 For example, while proteinuria at biopsy between 1.0–1.5 g/day with M0 and T0 has a 90.6% risk of composite renal outcome, for those with M1 (mild) and T1 or T2 (mild or moderate), the risk is 73.5%.13
To illustrate this, De Vriese discussed two cases which, on first examination may appear similar; both were 45-year-old males with proteinuria levels of 1.5 g/day (Figure 1). “From the perspective of the KDIGO guidelines,” said De Vriese, “you would need to approach these patients in a similar way. However, the first patient does not have persistent microscopic haematuria, nor does he have any signs of active disease in the kidney biopsy, while the second patient does.”4
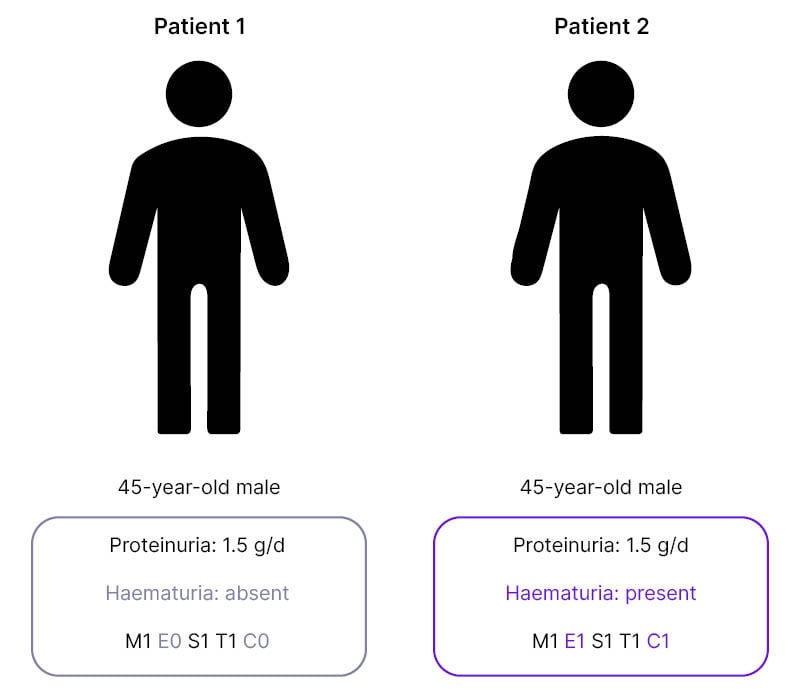
Figure 1: Two cases of IgA nephropathy with differences in haematuria and MEST-C score.
C: crescents; E: endocapillary hypercellularity; M: mesangial hypercellularity; S: segmental glomerulosclerosis; T: tubular atrophy/interstitial fibrosis.
This means, De Vriese continued, that “these patients have a different risk of progressive disease, and it’s highly unlikely that this first patient will respond to a course of immunosuppressive drugs.”
Current Treatment for IgA Nephropathy
As immunosuppressant agents are currently the main treatment choice when optimised supportive care does not reduce proteinuria, a handful of trials have been carried out to assess the utility of this approach. In the ‘Therapeutic Evaluation of Steroids in IgA Nephropathy Global’ (TESTING) trial, 6−9 months’ treatment with methylprednisolone (n=257) was compared with placebo (n=246); both study groups included optimised supportive care.14 Over the 6-year trial and follow-up period, cumulative incidence of the primary endpoint (40% decline in eGFR, kidney failure, or death due to CKD) was significantly lower (p<0.001) in the methylprednisolone group compared with placebo (hazard ratio: 0.53; 95% confidence interval [CI]: 0.39–0.72].14 However, De Vriese pointed out, supplementary data showed that following initial proteinuria decline during the active treatment period in the methylprednisolone group, proteinuria levels rose during the follow-up period. Conversely, eGFR levels initially rose with methylprednisolone treatment, but the slope of decline following discontinuation was similar to that of the placebo group.14
In the 3-year ‘Supportive Versus Immunosuppressive Therapy for the Treatment of Progressive IgA Nephropathy’ (STOP-IgAN) trial, 6 months’ glucocorticoid treatment combined with optimised supportive care was compared to optimised supportive care alone. At the 6-month timepoint, there was a decrease in proteinuria in the combined treatment group; however, once treatment was withdrawn, any advantages over optimised supportive care alone were not sustained at 36 months. At all timepoints, reported de Vriese, there was no beneficial effect with combined therapy on eGFR.1
The ‘Effect of Mycophenolate Mofetil on Renal Outcomes in Advanced Immunoglobulin A Nephropathy’ (MAIN) trial compared the immunosuppressive agent mycophenolate mofetil (MMF; n=85) with optimised supportive care (n=85) for at least 18 months, with a follow-up period of up to 3 years from baseline. At the end of the trial, primary composite endpoint outcomes (doubling of serum creatinine levels, kidney failure, or death due to kidney or cardiovascular cause) were significantly different between the groups (log-rank p=0.008).16
De Vriese noted though, that while the majority of MMF group participants discontinued treatment at the end of the trial phase (n=66), 14 MMF group participants remained on treatment throughout the post-trial follow-up, and differences were found in eGFR and proteinuria, according to whether or not participants discontinued or remained on treatment. Over the entire study period, in the optimised supportive care only group, eGFR steadily decreased from around 50 mL/min/1.73 m2 to around 30 mL/min/1.73 m2. In the MMF group, eGFR declined to around 47 mL/min/1.73 m2 by the end of the initial trial period; this was retained throughout the post-trial period in the group that continued taking MMF. However, eGFR declined to levels similar to the optimised supportive care group in the group that discontinued MMF use. For proteinuria, percentage decrease from baseline was significantly greater in the MMF group compared with the optimised supportive care group (p<0.001). At the end of the post-trial period, gains over the optimised supportive care group disappeared in the MMF group that discontinued treatment, but were retained in those who continued treatment.16
One drug highlighted in this session was a delayed-release formulation of the corticosteroid budesonide,17-19 which has received conditional approval from the European Medicines Agency (EMA),20 and accelerated approval from the U.S. Food and Drug Administration (FDA)21 for adults with IgAN at high risk of progressive disease (proteinuria >1.5 g/day). This drug, explained De Vriese, “has been designed to selectively affect the immune cells in the gut, located in the Peyer’s patches, that are thought to be responsible for Gd-IgA1 and anti-Gd-IgA1 autoantibodies generation.”17,20,22,23 Budesonide has been formulated into triple-coated beads inside an enteric coated starch capsule. Together, they provide delayed-release only once the capsule is exposed to the pH of the intestine (around pH 5−8),17,20 in the area where Peyer’s patches are most abundant, the final 25 cm segment of the distal ileum.2 As only a small percentage of the active drug reaches the systemic circulation, compared to systemic glucocorticoids, it is postulated to have a lower toxicity profile.17,20
The ‘Efficacy and Safety of Nefecon (delayed-release budesonide) in Patients with Primary IgA Nephropathy’ (NefIgArd) study was a Phase III trial that compared delayed-release budesonide (n=182) with placebo (n=182) in patients with IgAN and persistent proteinuria, all of whom received optimised supportive care.25,26 Results showed that over 9 months of treatment, mean change from baseline in eGFR in the delayed-release budesonide group was 0.66 mL/min/1.73 m2 (95% CI: -0.80–2.15) compared with -4.56 mL/min/1.73 m2 (95% CI: -5.86–-3.22) in the placebo group. Mean change from baseline in proteinuria was -33.6 (95% CI: -39.6–-27.0) in the delayed-release budesonide group compared with -5.2 (95% CI: -13.7–4.3) in the placebo group.24 However, noted De Vriese when examining the observational follow-up period, “after withdrawal of budesonide, proteinuria slowly starts to creep up again and, after an initial rise in eGFR, the slope of decline runs in parallel with the placebo group.”25
With all of the studies of glucocorticoids in mind, De Vriese noted that, in simple terms, they showed that “glucocorticoids work for as long as you give them, and when you stop giving them, you’re back to where you came from.”
Another treatment discussed in this session was the addition of a sodium/glucose cotransporter-2 (SGLT-2) inhibitor to RAAS inhibitors. SGLT-2 inhibitors enhance urinary glucose excretion by reducing renal proximal convoluted tubule glucose reabsorption, and have an effect on efferent arteriolar tone. As SGLT-2 inhibitors affect afferent arteriolar tone, the combination of these two agents can reduce intraglomerular pressure in an additive manner.25,26
New Treatment Opportunities in IgA Nephropathy
A number of agents are being trialled that target IgAN along different stages of its four-hit pathogenesis. These include agents that target the kidney, including AT1 and ETA blockers; those that block production of Gd-IgA1 and anti-Gd-IgA1 autoantibodies in the gut through B-cell/antibody suppression; as well as complement inhibitors (Table 1).9 De Vriese discussed how now, or shortly, “for IgAN pathogenesis, there is budesonide and other drugs in development [such as the] anti-a proliferation-inducing ligand and/or anti-B-cell activating factor, and anti-CD38 inhibitors/antibodies. For inflammation you have the systemic immunosuppressants and complement inhibitors, and for fibrosis you have RAAS inhibition and sparsentan.”
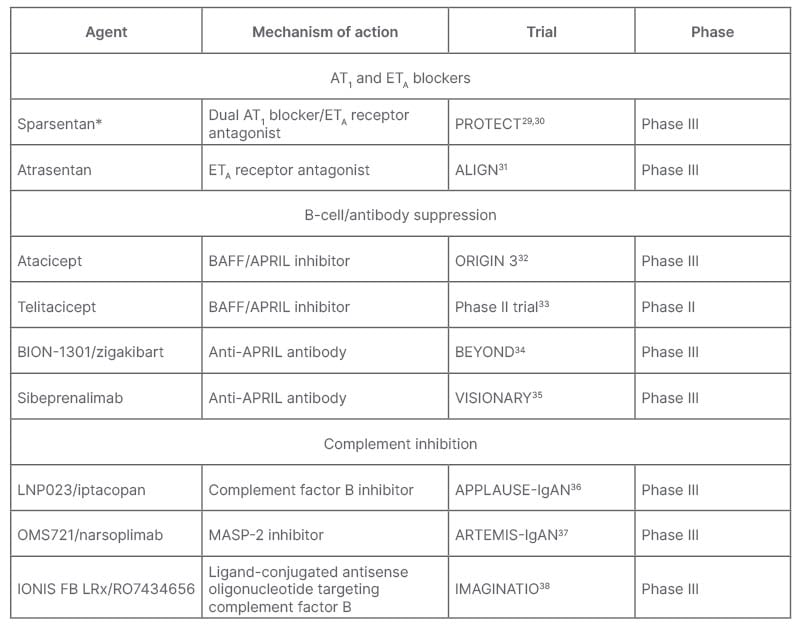
Table 1: Agents in development or newly approved for treatment of IgA nephropathy.9
*Recently received FDA accelerated approval.39
APRIL: a proliferation-inducing ligand; AT1: angiotensin II receptor type 1; BAFF: B-cell activating factor; ETA: endothelin receptor type A; FDA: U.S. Food and Drugs Administration; IgAN: immunoglobulin A nephropathy; MASP-2, mannan-binding lectin serine protease 2.
Sparsentan for IgA Nephropathy
Optimised supportive care, discussed Tesař, may soon include drugs that target the endothelin receptor, as such agents are currently in Phase III trials for IgAN.29-31 The vascular endothelin system is comprised of endothelins-1, -2, and -3. Receptors for ET-1, primarily ETA but also ETB, are both expressed in the kidneys. ET-1 has a variety of effects on vascular and renal cells, and is associated with CKD pathophysiology. For example, in the vasculature, ET-1, via ETA receptors, contributes to vasoconstriction, platelet activation, and endothelial dysfunction, with overexpression leading to hypertension and/or atherosclerosis. In the kidney, ET-1 actions, also via ETA receptors, include those on podocyte-related proteinuria, cytoskeletal remodelling, and foot process effacement; in mesangial cells, ET-1 has a role in glomerulosclerosis, extracellular matrix accumulation, and cell hypertrophy; and in renal tubules, ET-1 is linked to tubulointerstitial fibrosis. These actions may all lead to CKD.40
With the actions of ET-1 in mind, it is of interest for IgAN treatment that ETA blockade can lead to reductions in proteinuria, blood pressure, and salt and water retention, along with decreases in reactive oxygen species, fibrosis, and inflammation. Such blockade is associated with reductions in hypertension, proteinuria, and diabetic micro- and macrovascular complications.39 Studies of ETA and ETB receptor antagonists show that while some drugs, such as avosentan, are non-ET-1 receptor selective, others, including sparsentan and atrasentan, are much more selective for ETA.42
There are a number of interactions between the ET-1 system and the RAAS. For example, while angiotensin II can stimulate ET-1 release in renal cells43 and enhance ET-1-induced vasoconstriction,44 ET-1 can stimulate aldosterone secretion, inhibit juxtaglomerular apparatus renin release, and stimulate angiotensin II formation in pulmonary endothelial cells.43 Sparsentan is a first-in-class dual ETA and AT1 receptor antagonist that can bind to these receptors in podocytes, mesangial cells, and other renal cell types, preventing binding of ET-1 and angiotensin II, respectively.43,45,46 It recently received accelerated approval by the FDA39 in the USA and marketing authorisation is currently being evaluated in Europe. Studies evaluating sparsentan have shown that it can prevent actions such as vasoconstriction and endothelial dysfunction in renal vessels; glomerulosclerosis, mesangial cell proliferation, and extracellular matrix production in the glomeruli; and proteinuria, podocyte apoptosis, and increased glomerular permeability, that can lead to inflammation and fibrosis, in the tubulointerstitial compartment.43,47
In the Phase III, international, multicentre, randomised, double-blind, active-controlled PROTECT trial, 400 mg/day sparsentan (n=202) was compared with maximum dose (300 mg/day) of the AT1 blocker irbesartan (n=202) in adult participants with IgAN and persistent proteinuria, despite maximum angiotensin-converting enzyme inhibitors or AT1 blockers.29,30 Patients were predominantly male (70%), White (67%), and had a history of hypertension (70%). The initial double-blind trial period was for 110 weeks, followed by 4 weeks of standard of care without study medications. There is an ongoing, optional, open-label extension period of up to 156 weeks. The primary efficacy endpoint was change in urinary protein-to-creatinine ratio (UP/C) at Week 36. Secondary endpoints include the rate of change in eGFR from baseline to 110 weeks, as well as eGFR rate of change following the initial acute effects of randomised therapy, that is, from Week 6 to Week 58, and to Week 110.29,30
In the interim analysis at Week 36, there was a -49.8% (95% CI: -55.0–-44.0) change in UP/C for the sparsentan group compared to a -15.1% (95% CI: -23.7–-5.4) change in the irbesartan group. This represented a 41% relative reduction in UP/C ratio and a significant difference, favouring sparsentan (p<0.0001). Significant differences were shown as early as Week 4, with an initial large change in UP/C in participants receiving sparsentan, followed by further decreases over the trial period.30
Also observed at Week 36 was complete proteinuria remission (urinary protein excretion <0.3 g/day) in 21% of participants in the sparsentan group, and 8% of participants in the irbesartan group (odds ratio: 3.1; 95% CI: 1.6–5.8; p=0.0005). Partial proteinuria remission (urinary protein excretion <1.0 g/day) was observed in 70% of participants in the sparsentan group, and 44% of participants in the irbesartan group (odds ratio: 4.5; 95% CI: 2.7–7.6; p<0.0001). Baseline subgroup analysis, assessing several variables, such as sex, eGFR level, urine protein, and age, showed that higher proteinuria reductions were consistent across subgroups. Tesař also observed that there were limited differences between the two treatment groups with regard to blood pressure,30 meaning that proteinuria remission was not solely caused by the effect of sparsentan on blood pressure.
Treatment emergent adverse events in the sparsentan and irbesartan groups were similar, and included peripheral oedema (14% and 9% of participants, respectively), hypotension, (14% and 6%), hyperkalaemia (13% and 10%), and dizziness (13% and 5%). Treatment emergent adverse events leading to treatment discontinuation were reported in 7% of participants in the sparsentan group and 5% of participants in the irbesartan group. New diuretic use occurred in 18% of participants in the sparsentan group and 19% of participants in the irbesartan group. No participant in either group discontinued treatment due to oedema, there was no evidence of heart failure, and there were no serious adverse events associated with treatment-related fluid retention. Where liver enzyme elevations >3x the upper limit of normal occurred (2% of participants in each group), they were asymptomatic, reversible, and without concurrent elevation in total bilirubin.30,48
Tesař concluded with the reminder that “even with all these treatments we now have available, still a significant proportion of patients progress to kidney failure, partly because they can’t take a medication that may work for a long time due to adverse events. Based on the interim analysis of the PROTECT trial, it seems that combined inhibition of AT1R and ETAR, is a promising strategy, although we need to have the data from the full study to see if the effect on proteinuria is translated to a beneficial eGFR effect.”
Conclusion
A significant proportion of patients with IgAN show a progressive disease course. Current treatment recommendations primarily include RAAS inhibition, immunosuppressant treatment, and lifestyle alterations, but a ‘one size fits all’ approach may not target all symptoms in every patient.1,2,4 Delayed-release budesonide17-19 and the dual ETA and AT1 receptor antagonist sparsentan,43-46 which recently received accelerated FDA approval,39 are now available in some countries for people with progressive IgAN, and there are several other drugs in development. The availability of such drugs should mean that each of the four hits of IgAN pathogenesis can now be targeted, depending on the individual symptoms of each patient.