Abstract
This case report highlights a rare and unique occurrence: the simultaneous presence of sickle cell thalassaemia and overlapping syndrome, which involves systemic lupus erythematosus and autoimmune hepatitis. The coexistence of sickle cell disease with overlapping syndrome is exceptionally rare, with only a few documented cases in the literature, one of which involves sickle cell β-thalassaemia. Significantly, this case enhances understanding of the intricate relationship among these conditions, and offers valuable perspectives on how to clinically manage them. The authors present the case of a young male in his early 20s, who presented with haemolytic anaemia, jaundice, joint pain, and hepatomegaly. Extensive laboratory investigations, including serological markers, haemoglobin electrophoresis, and liver function tests, confirmed the coexistence of sickle cell thalassaemia, systemic lupus erythematosus, and autoimmune hepatitis. The treatment included O2 therapy, hydration, hydroxyurea, and antibiotics. After 4–5 days, the patient showed improvement, and at discharge, hydroxyurea and folic acid were continued. Significantly, considering the complex medical history of the patient, a decision was made to include a carefully considered, low-dose steroid regimen. The choice of a maintenance dose over an induction therapy was specifically made to mitigate potential complications, particularly the risk of vaso-occlusive crises in patients with sickle cell disease. This case report contributes to the understanding of concurrent manifestation of these complex conditions, and emphasises the importance of a comprehensive approach, early diagnosis, and timely management, to optimise patient outcomes in such intricate overlapping syndromes.
Key Points
1. Importance of recognising overlapping symptoms: this case report emphasises the critical role of physicians in identifying overlapping symptoms, particularly in patients with sickle cell thalassaemia. Recognising the coexistence of autoimmune illnesses is critical for early management and improved patient outcomes.2. Intricate interplay between sickle cell disease and autoimmunity: the case report discusses the complex relationship between sickle cell disease and autoimmune illnesses, emphasising the elevated risk and clinical implications. Elevated antinuclear antibody levels, possible problems from hydroxyurea, and the nuanced use of steroids highlight the various issues in managing these individuals.
3. Diagnostic complexity and a call for research: the case report emphasises the challenges in diagnosing concurrent autoimmune diseases in patients with sickle cell, due to overlapping clinical characteristics. Comprehensive assessments are required, and the article calls for more research to uncover the underlying processes. This approach seeks to improve diagnosis accuracy, and enable more effective treatment solutions.
INTRODUCTION
Sickle cell disease (SCD) is a genetic condition caused by mutations in the haemoglobin (Hb) gene, resulting in various complications. One specific form of SCD is sickle cell thalassaemia, which occurs by inheriting both sickle cell and thalassaemia traits.1,2 SCD encompasses several other variants, including sickle cell trait, sickle cell anaemia, and sickle cell Hb-C disease, each with distinct characteristics.3,4 Systemic lupus erythematosus (SLE) is a complex autoimmune disease characterised by autoantibodies binding to tissues, forming immune complexes, and causing damage across various organs, leading to chronic inflammation.5 The disease is more prevalent in females, with a female-to-male ratio of 9:1.6 This condition arises when the immune system mistakenly targets healthy cells and tissues, leading to a diverse array of symptoms, including joint pain, fever, fatigue, skin rashes, and potential damage to vital organs, such as the kidneys, heart, and lungs.7 Autoimmune hepatitis is a chronic liver disease in which the immune system mistakenly attacks liver cells, leading to inflammation and potential damage.8
While SCD has been extensively studied, conditions such as sickle cell thalassaemia and autoimmune disorders that overlap with it remain relatively rare, with only a limited number of reported cases, including a scarcity of sickle cell β-thalassaemia cases.2,8 Although sickle cell thalassaemia and SLE are distinct medical conditions with different underlying pathophysiological mechanisms, they can present with overlapping symptoms, such as anaemia, fever, joint pain, arthritis, and chronic inflammation.1,9 This overlap in clinical manifestations can often lead to difficulties and delays in the diagnosis of SLE in patients with SCD.3 Furthermore, individuals with SCD have compromised activation of the alternate complement pathway, making them more susceptible to infections, and potentially prone to autoimmune disorders.4,9 Patients with SCD may also experience hepatic crises due to red blood cell sickling, leading to hepatomegaly, elevated liver enzyme levels (serum transaminases), or cholestasis.10 Liver issues in these patients can result from various factors, including hepatic sequestration, viral hepatitis from blood transfusions, haemosiderosis (iron accumulation in the liver), gallstones, or unrelated medical conditions.10,11 However, an exceedingly rare association has been observed between sickle cell thalassaemia and autoimmune hepatitis, with only a handful of published cases documented.8,11
In this case report, the authors present a case of the coexistence of SLE and autoimmune hepatitis in a male patient with sickle cell thalassaemia. The occurrence of this uncommon overlapping syndrome highlights the complexities involved in diagnosing and managing these distinct conditions, emphasising the need for careful consideration and evaluation.
CASE PRESENTATION
Patient Information
In this case report, the authors present the admission of a male in his early 20s from the rural interior Sindh region of Pakistan. The patient exhibited symptoms over a 3-day period, including yellowing of the sclera, darkening of urine, pale stools, itching, fever, and joint pain. Notably, there were no known comorbidities, or a family history of similar conditions.
Clinical Findings
During the physical examination, the patient had a normal body temperature of 36.7 °C, blood pressure of 110/70 mmHg, and a heart rate of 88 beats/min. He showed signs of anaemia, moderate jaundice, an elevated jugular venous pressure, a facial rash, a receding hairline, and an enlarged liver located 2 cm below the right costal margin, with sharp margins, firm consistency, and no tenderness. He had pain on pressure in the long bones, and knee arthritis with normal joint movements. This patient previously had a similar episode of eye yellowing, and received red blood cell transfusions a few years ago. In the last year, he experienced a recurrence, which necessitated four units of packed red blood cell transfusion and a bone marrow biopsy. He did not use any routine medications, such as antimalarial prophylaxis, steroids, or pain relievers.
Diagnostic Assessment
Extensive investigations revealed findings contributing to the patient’s differential diagnosis. The malaria panel revealed positive results for both the malarial parasite test and the malarial parasite immunochromatographic test, confirming the presence of Plasmodium vivax. Blood test results showed normocytic anaemia (Hb: 5.8 g/dL) with a corrected reticulocyte count of 2.53%, hyperleukocytosis (leukocyte count: 37,000 cells/mm³), and a platelet count of 156,000 cells/mm³. The peripheral film showed the malarial vivax trophozoites and gametocytes, along with anisocytosis, poikilocytosis, polychromasia, target cells, fragmented red blood cells, nucleated red blood cells, and sickle cells. Additionally, the sickling test was positive, indicating sickle cell thalassaemia. Hb electrophoresis further confirmed this diagnosis, with Hb S at 95.6% and Hb A2 at 4.40%. The diagnostic evaluation of the patient revealed significant findings indicative of SLE.
Immunological investigations demonstrated a positive antinuclear antibody (ANA) titre of 1/2,560, with a coarse speckled pattern. The anti-double-stranded DNA antibody was elevated at 30.8, indicating the presence of antibodies against double-stranded DNA. Further positive results included SS-A/Ro antibodies at 50.11, and while serum C3 levels were low, C4 and CH50 levels, as well as thyroid and renal functions, were all within acceptable limits. The elevated levels of inflammatory markers, such as C-reactive protein (88.3 mg/dL), erythrocyte sedimentation rate (30 mm/hour), ferritin (20,000 ng/mL), and lactate dehydrogenase (1,168 U/L), further suggested an active inflammatory process, consistent with SLE. The diagnostic workup also revealed evidence supporting a diagnosis of autoimmune hepatitis. The immune laboratory tests showed a positive ANA, with a coarse speckled pattern at a titre of 1/2,560, indicative of autoimmune involvement. Additionally, anti-smooth muscle antibodies tested positive. Serum IgG levels were elevated at 28.74 g/L, surpassing the normal reference range of 6.12–16.16 g/L. Liver function tests displayed total bilirubin elevated at 9.5 mg/dL, direct bilirubin at 6.0 mg/dL, and indirect bilirubin at 3.5 mg/dL, suggesting a pattern of hyperbilirubinaemia indicative of hepatic dysfunction. Transaminases were significantly elevated, with aspartate aminotransferase at 244 IU/L, and alanine transaminase at 129 IU/L, surpassing the normal reference ranges. Alkaline phosphatase was elevated at 158 IU/L, and serum γ-glutamyl transferase was elevated at 193 IU/L, indicating cytolysis and cholestasis (Table 1).
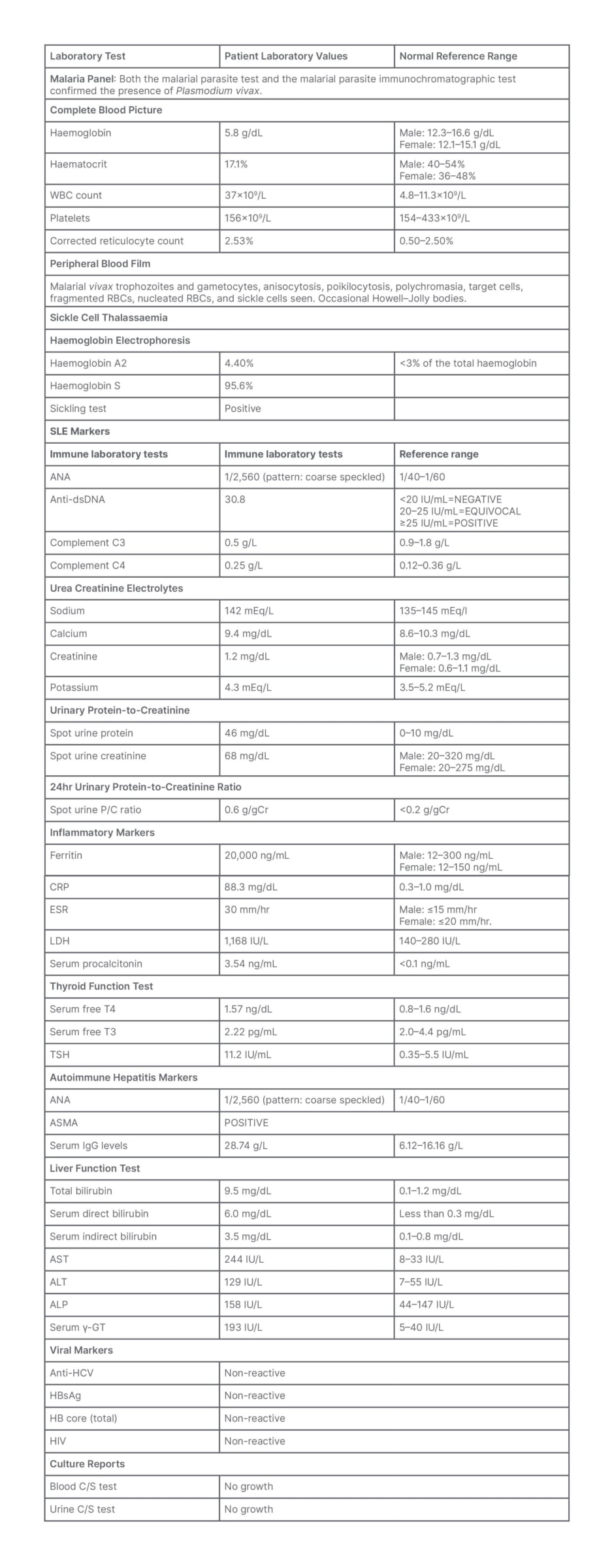
Table 1: Patient blood panel.
ANA: antinuclear antibody; ALP: alkaline phosphatase; ALT: alanine transaminase; ASMA: anti-smooth muscle antibody; AST: aspartate aminotransferase; CRP: C-reactive protein; C/S: culture and sensitivity; dsDNA: double-stranded DNA; ESR: erythrocyte sedimentation rate; HBsAg: hepatitis B surface antigen; HCV: hepatitis C virus; LDH: lactate dehydrogenase; RBC: red blood cell; serum γ-GT: serum γ-glutamyl transferase; SLE: systemic lupus erythematosus; TSH: thyroid-stimulating hormone; WBC: white blood cell.
Chest radiography and echocardiography results were normal. Abdominal ultrasonography revealed an enlarged liver with a span of 15.7 cm, smooth margins, and altered echotexture. Rheumatoid factor, anti-U1-ribonucleoprotein antibodies, anti-SS-B/La antibodies, anti-Smith antibodies, anti-Scl-70 antibodies, and anti-cyclic citrullinated peptide antibodies were all negative, as was viral serology. Tests for anti-phospholipid and anti-thyroid peroxidase antibodies were negative. The results of the Coombs test and glucose-6-phosphate dehydrogenase test were negative.
In the bone marrow biopsy conducted 1 year ago, all three primary cell lines were observed. Notably, there was a slight increase in the production of red blood cells, which displayed a typical maturation process. The myelopoiesis, responsible for white blood cell formation, exhibited a balanced ratio of 1:2. The percentage of blast cells (immature blood cells) was less than 4%, while plasma cells constituted 2% of the total cell population. Furthermore, the biopsy identified the presence of megakaryocytes (involved in platelet production), and featured prominent eosinophilic precursors (immature eosinophils, a specific type of white blood cell).
On the second day of hospital admission, the patient complained of relative constipation followed by abdominal distension and discomfort. Upon examination, the abdomen was tense and tender, with fullness around the umbilicus, and decreased gut sounds were noted upon auscultation. In addition, during a digital rectal examination, ballooning of the rectum was observed. Considering the possibility of acute abdominal obstruction, urgent abdominal X-ray and ultrasound were performed. The imaging studies shown in Figure 1A and 1B revealed multiple air-fluid levels and dilated bowel loops on the left side of the abdomen, indicating the presence of an obstruction. Furthermore, peristaltic movements of the bowel were observed to be sluggish on ultrasound. Due to the severity of the condition, the surgical team was consulted. In addition to the abdominal symptoms, the patient developed a high spiking fever and a continuously rising total leukocyte count. The patient was in a sickle crisis, which explained the presenting symptoms of constipation, abdominal distention, and fever. A CT scan was planned; however, it had to be deferred due to the patient’s declining medical condition at the time. Subsequently, following the receipt of laboratory results, a liver biopsy was proposed as a means to establish a conclusive diagnosis of autoimmune hepatitis. Regrettably, the patient declined to provide consent for the liver biopsy procedure.
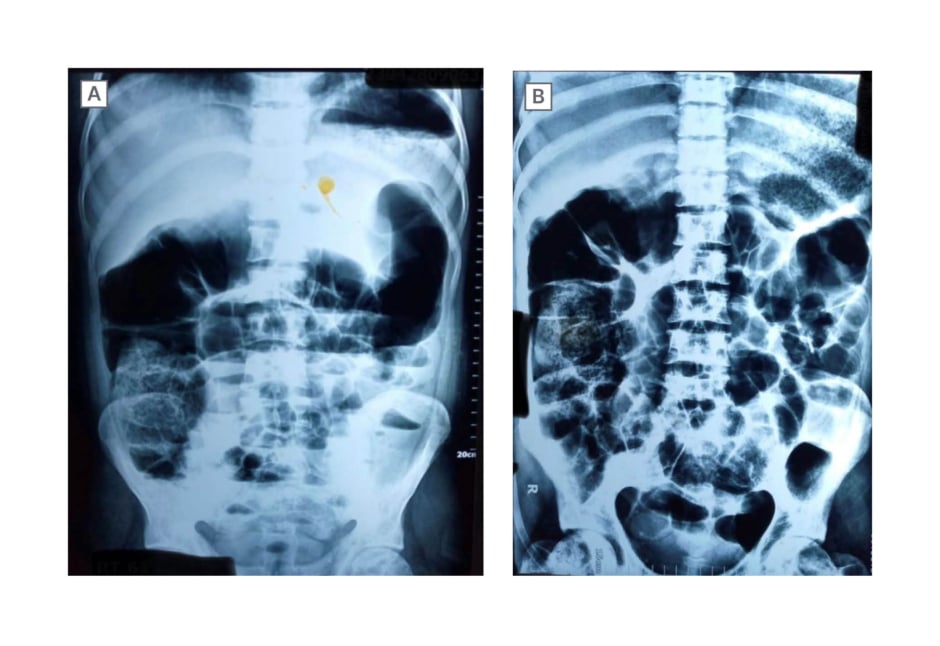
Figure 1: Patient abdominal X-ray.
These figures present anteroposterior projection abdominal X-ray images taken in the supine position. The images reveal multiple air-fluid levels and dilated bowel loops on the left side of the abdomen, indicating the presence of an obstruction.
During the patient’s follow-up appointment, a 24-hour urinary protein-to-creatinine ratio test was performed, and the results fell within the established normal range, affirming normal renal function without evidence of autoimmune-related complications. Notably, spot urine parameters, including a protein concentration of 46 mg/dL and creatinine level within reference ranges, supported this finding. The spot urine protein-to-creatinine ratio of 0.6 g/gCr, above the threshold of 0.2 g/gCr, further confirmed the absence of significant renal issues. The results provide valuable insights into effective kidney filtration and regulation of protein excretion. It is important to note that an elevated spot urinary protein-to-creatinine ratio, as observed previously, can be influenced by factors such as infection, dehydration, stress, and specific dietary choices. These normal findings contribute to a comprehensive assessment, reassuring the absence of autoimmune-related renal complications. Ongoing monitoring was advised for continued care.
Therapeutic Intervention
The treatment regimen encompassed a multifaceted approach to manage the sickle crisis. O2 therapy was provided to enhance O2 saturation, intravenous fluids were administered to maintain proper fluid balance, and hydroxyurea was initiated at a daily dose of 500 mg for its disease-modifying properties. The patient received periodic packed red blood cell transfusions to address diminishing haemoglobin levels, but no phlebotomy was performed. Furthermore, the patient was prescribed meropenem 1 g intravenously three times daily to address potential infections, and a course of Gen-M (120 mg intravenously) was administered for the treatment of malaria. In light of the presence of other autoimmune diseases, and a decline in complement levels indicative of a disease flare-up, the patient was started on a low-dose steroid regimen, ranging from 10–20 mg per day, followed by a tapering schedule. The decision to incorporate steroids into the treatment plan was carefully considered, taking into account the patient’s intricate medical history. However, in an effort to mitigate potential complications, particularly the risk of vaso-occlusive crises in patients with SCD, a maintenance dose of steroids was chosen over an induction therapy. Importantly, no immunosuppressive agents were introduced during the course of treatment. This careful choice was made to keep a careful balance in taking care of the different parts of the patient’s health, without adding extra factors that might influence the authors’ understanding of the treatment results.
Follow-up and Outcomes
Following the 4–5-day treatment period, the patient showed substantial improvement in symptoms. Pain became less severe and less frequent, the fever resolved, and relief from constipation and abdominal distention was observed. Upon discharge, the patient’s treatment plan included the continuation of hydroxyurea and folic acid, along with a tapering dosage schedule of steroids. Despite the initial challenges, the patient’s recovery was uneventful, with no observed complications or recurrence of symptoms after treatment.
DISCUSSION
This case report provides a comprehensive analysis of a male patient concurrently diagnosed with sickle cell thalassaemia, SLE, and autoimmune hepatitis. It sheds light on the intricate interplay between these conditions, prompting crucial considerations for further exploration.1,3 When patients with sickle cell thalassaemia exhibit unusual clinical characteristics or multi-organ involvement, clinicians must consider overlapping syndromes.2,9
While the exact mechanisms responsible for these interactions are not fully understood, it is hypothesised that individuals with these interactions may possess an aberrant alternate pathway within their complement system, potentially heightening their vulnerability to autoimmune disease development.4,9 Another hypothesis posits that individuals afflicted with this condition may have an immune system that does not function optimally. This immune dysfunction may be attributed to factors such as diminished spleen function, disruptions within the complement pathway, and compromised processes of opsonisation and phagocytosis, contributing to the pathogenesis. As a consequence, the effective clearance of immune complexes may be compromised. It has been suggested that there is suspicion of SLE in a patient with SCD.12 The clinical suspicion is supported by the finding that patients with SCD tend to have higher ANA titres than the general population.13
In addition, it is hypothesised that SLE is the most commonly reported autoimmune disorder in SCD.3,13,14 The coexistence of SCD and autoimmune hepatitis is also uncommon, with only a few documented cases in the literature. Autopsy studies suggest a prevalence of cirrhosis ranging from 16–29% in patients with sickle cell anaemia. The main contributing factor for liver disease in individuals with SCD is receiving multiple blood transfusions, which is associated with infections such as hepatitis B virus (HBV) and hepatitis C virus, along with the accumulation of excessive iron in the liver.15,16 In an important study, it was observed that children who got the HBV vaccine had a significant drop in HBV infections for 3–5 years after getting vaccinated. This highlights the effectiveness of the vaccination in reducing the incidence of HBV among at-risk groups, emphasising the positive impact of timely vaccination measures in preventing infections.17 Considering the heightened risk, the patient was also counselled to undergo HBV vaccination during subsequent follow-up appointments. Nevertheless, the presence of ANA positivity at a titre of 1/160 may indicate the potential existence of concurrent autoimmune hepatitis as a plausible explanation for liver damage.16
Managing the presence of both autoimmune diseases and SCD is a challenging task. Decisions regarding overlap syndromes should be carefully made under specialised guidance to optimise outcomes and minimise risks, especially in individuals with SCD.18 Hydroxyurea, a frequently used treatment for SCD, plays a pivotal role in reducing the frequency and severity of complications associated with SCD, including painful events, preservation of splenic function, and attenuation of sickle-related organ damage.19 This drug suppresses the production of autoantibodies, potentially concealing early autoimmune disease.20 As a matter of fact, corticosteroids are an appealing treatment for severe SCD complication, because they are inexpensive, readily available, and exhibit strong anti-inflammatory effects.21
In this particular situation, a decision was made to refrain from using full-dose steroids, despite their known anti-inflammatory properties. This decision was made by evidence suggesting an elevated risk of vaso-occlusive crises and subsequent hospitalisation in patients with SCD who undergo steroid treatment.22 Several studies suggest that treatments, such as those targeting TNF, may offer potential benefits for autoimmune diseases associated with SCD.23 Immunosuppressive and anti-inflammatory medications have the potential to cause kidney and liver damage, which could be exacerbated by complications related to SCD. They also heighten the susceptibility to infections.24 The commencement of such therapies should be a subject of consultation at specialised centres, and calls for vigilant oversight.25 Given the overlapping clinical features and the high prevalence of positive ANA in individuals with SCD, clinicians encounter significant challenges in accurately identifying coexisting autoimmune disorders.4 A delay in diagnosis contributes to elevated morbidity and mortality associated with both autoimmune diseases and SCD.25
This case report underscores the pressing need for further research to deepen the understanding of the pathophysiology, natural history, and optimal treatment approaches for overlapping syndrome and sickle cell thalassaemia. In addition, the necessity of a multidisciplinary approach, encompassing medical specialists such as haematologists, rheumatologists, and nephrologists, is imperative due to the involvement of multiple organ systems in these coexisting conditions. Effective treatment demands the collective expertise of these specialised medical professionals. Continued investigation is imperative to advance patient care and outcomes in this complex clinical scenario.
CONCLUSION
High Index of Suspicion for Overlapping Syndromes
This report illustrates the unique occurrence of concomitant manifestation of sickle cell thalassaemia with SLE and autoimmune hepatitis, representing a complex and intriguing overlapping syndrome. Clinicians should be alert for signs of competing conditions while treating patients with sickle cell thalassaemia with unusual symptoms, or significant organ dysfunction. The coexistence of these illnesses highlights the importance of investigating any additional problems.
Sickle Cell Disease and Autoimmune Diseases
Patients with SCD are more likely to develop autoimmune disorders. Elevated ANA levels in these patients may raise clinical concerns about conditions such as SLE, prompting more research into the underlying processes and unusual complement pathways.
Hydroxyurea and Autoimmune Disorders
Hydroxyurea, a drug commonly used for the treatment of SCD, suppresses the production of autoantibodies, and may mask the early signs and symptoms of autoimmune conditions. Healthcare workers should be aware of this effect.
Use of Steroids in Sickle Cell Disease
Due to the increased risk of vaso-occlusive crises and hospital admissions associated with their use, the choice to use steroids in patients with SCD is frequently avoided, even though they possess recognised anti-inflammatory properties.
Challenges in Identifying Coexisting Autoimmune Disorders
The similarity in clinical features and high frequency of patients with SCD who are ANA-positive make it difficult for clinicians to diagnose between concurrent autoimmune diseases. The diagnostic journey for this patient involved comprehensive laboratory investigations, serological markers, and imaging studies to confirm the presence of these overlapping disorders. To understand the underlying mechanisms causing the coexistence of these rare illnesses, and to investigate other management strategies, more research is required. Increased awareness and knowledge of such overlapping syndromes will contribute to improved diagnosis, treatment, and overall care for patients facing similar challenges.
Patient Perspective
Living with multiple illnesses can be incredibly challenging. Some days, you experience intense pain and constant fatigue, making it hard to make plans due to the unpredictability of getting sick. Frequent doctor visits become a routine. However, despite the hardships, you grow stronger, and learn to cherish the positive moments in life. It is a journey filled with numerous obstacles, ultimately shaping you into a more resilient individual.
Note: This perspective was originally shared by the patient in another language and has been translated by the doctor for reference.






