Abstract
Antiphospholipid syndrome (APS) is an acquired form of thrombophilia characterised by the presence of antiphospholipid antibodies and arterial/venous thrombosis or obstetric complications. Although antiphospholipid antibodies are reported in 1–5% of the general population, only a minority of these individuals will develop the clinical manifestations of APS. The typical expressions of APS are thrombotic events that can involve veins, arteries, or small vessels in any organ or tissue. Pregnancy morbidity refers mainly to early and late fetal loss, but pre-eclampsia, eclampsia, or placental insufficiency can also occur. Extra-criteria manifestations include thrombocytopenia, APS-associated nephropathy, valvular heart disease, neurological manifestations, and livedo reticularis. The diagnosis of APS is currently based on the Sydney criteria: i.e., meeting at least one clinical criterion (vascular thrombosis or pregnancy morbidity) and one laboratory criterion (lupus anticoagulant, anticardiolipin antibodies, or anti-β2 glycoprotein-I antibodies). Anticoagulation with unfractionated or low molecular weight heparin followed by vitamin K antagonist is the standard treatment for APS patients presenting with venous thromboembolism. There is not enough evidence regarding the use of the direct oral anticoagulants in this population. Patients presenting with arterial thrombosis may receive a combination of vitamin K antagonists and low-dose aspirin. In women with obstetrical APS, the combination of low molecular weight heparin and low-dose aspirin is usually prescribed to prevent pregnancy complications. The aim of this narrative review is to summarise the latest evidence on the diagnosis and antithrombotic treatment of APS.
Introduction
Antiphospholipid syndrome (APS) is an acquired form of thrombophilia with immune pathogenesis and is characterised by the presence of antiphospholipid antibodies (aPL) and clinical manifestations of arterial or venous thrombosis or obstetric complications. APS is sometimes known as Hughes syndrome, named after the author who first described a common pathogenic mechanism underlying recurrent venous thrombosis, cerebral diseases, and recurrent abortions in patients with systemic lupus erythematous (SLE).1
Epidemiology
Although aPL are reported in 1–5% of the general population,2 only a minority of these individuals will develop the clinical manifestations of APS. The incidence of APS is approximately 5 new cases per 100,000 people per year, while the prevalence is 40–50 cases per 100,000 people.3 The prevalence of aPL in patients with clinical events is higher: 13.5% in stroke, 11.0% in myocardial infarction, 9.5% in deep vein thrombosis, 6.0% in pregnancy morbidity, and 26.4% in women with recurrent early pregnancy loss.4,5
APS can occur without other conditions, known as primary APS, or can be associated with other autoimmune diseases, such as SLE or rheumatoid arthritis, which is known as secondary APS. The prevalence of aPL in SLE patients can reach 40%,2 and 20–50% of these will develop thrombotic events.6
Pathophysiology
aPL are autoantibodies directed against cell surface proteins bound to anionic membrane phospholipids. They are a heterogeneous group of autoantibodies, including lupus anticoagulant (LAC), anticardiolipin (aCL) antibodies, and anti-β2 glycoprotein-I (aβ2-GPI) antibodies.
The history of aPL dates back to the beginning of the 20th century, with the discovery of biological false-positive serological tests for syphilis, due to the interaction between aCL and cardiolipin used as a reagent in these assays.7 LAC was first described in the 1950s, in patients with SLE and prolonged clotting time in vitro, hence the name lupus anticoagulant.8 However, LAC can also be found in patients without SLE and it is known today as the paradox between the prolonged phospholipid-dependent coagulation tests in vitro and the hypercoagulable state in vivo.
The central role of antibodies against β2-GPI, a complement regulator and inhibitor of coagulation, was discovered in the 1990s,7 and a specific immunoassay for aβ2-GPI was developed. β2-GPI is the key antigen for all aPL. β2-GPI is a cofactor for aCL to bind cardiolipin, and the aCL that recognise the β2-GPI (β2-GPI-dependent aCL) correlate more strongly with thrombosis and obstetric complications, compared to β2-GPI-independent aCL.9 It was also demonstrated that LAC activity due to aβ2-GPI (β2-GPI-dependent LAC) is more correlated with thrombotic events than β2-GPI-independent LAC (such as LAC due to antiprothrombin antibodies).9,10 Furthermore, the aβ2-GPI can be specific for different domains of the β2-GPI molecule, and those antibodies directed towards the domain I were shown to be more predictive of clinical events.11
The pathophysiology of APS is still not completely understood. The main triggers for aPL synthesis are infections, due to the molecular mimicry between protein components of the infectious agents and cell surface proteins, such as β2-GPI.6 However, the presence of aPL alone (the ‘first hit’) is not sufficient to provoke a thrombotic event and a ‘multi-hit’ theory has been proposed, wherein other factors (such as infections, inflammatory diseases, surgery, immobility, and hormonal treatment) constitute the ‘second hit’ and drive the haemostatic balance towards thrombosis.6,8 Several pathways have been hypothesised to explain the procoagulant state induced by aPL, including complement activation, activation of platelets and endothelial cells, interference with the natural anticoagulants (protein C and tissue factor pathway inhibitor), and inhibition of fibrinolysis.8,12
Regarding obstetrical complications, the apoptotic effect of aPL on trophoblast cells can explain the early pregnancy morbidity (recurrent miscarriages), while ischaemic placental dysfunction can explain the late pregnancy morbidity: pre-eclampsia, intrauterine growth restriction or death, premature birth, and stillbirth.5
Clinical Manifestations
The typical manifestations of APS are thrombotic events that can involve veins, arteries, or small vessels in any organ or tissue. In the large cohort of 1,000 patients with APS enrolled in the Euro-Phospholipid Project,13 the most common clinical presentation was venous thromboembolism (VTE): deep vein thrombosis (31.7%), superficial thrombophlebitis (9.1%), and pulmonary embolism (9.0%). Arterial thrombosis (ATE) were less frequent: stroke (13.1%), transient ischaemic attack (7.0%), and myocardial infarction (2.8%).13 During the evolution of APS, a number of unusual-site thromboses were also reported, including cerebral vein thrombosis, mesenteric ischaemia, Budd–Chiari syndrome, renal artery or vein thrombosis, arterial thrombosis of the upper or lower extremities, and retinal artery or vein thrombosis.13,14
Pregnancy morbidity refers mainly to early and late fetal loss, reported in 35.4% and 16.9% of pregnancies in APS women, respectively.13 In the European Registry on Obstetric Antiphospholipid Syndrome, recurrent early miscarriage (53.8%) and late fetal loss (31.2%) were the most frequent obstetric complications.15 Other possible obstetric complications are pre-eclampsia, (9.5%), eclampsia (4.4%), and abruption placentae (2.0%).13
Furthermore, there are other clinical manifestations of APS, known as extra-criteria manifestations, which are not included in the classification criteria,16,17 but can be helpful to raise the suspicion of APS. The extra-criteria manifestations can be associated with thrombosis and pregnancy morbidity or can be isolated. Thrombocytopenia is reported in 20.0–46.0% of APS patients, is usually moderate (platelet count 50–100×103/mm3), and is associated more with thrombosis as opposed to bleeding risk.16,18 The APS-associated nephropathy is defined by the histopathologic finding of thrombotic microangiopathy, which can involve both arterioles and glomerular capillaries, and is closely correlated with kidney failure.18 Valvular heart disease includes sterile valve vegetations, thickening, and dysfunction, can lead to heart failure, and may require heart valve replacement.18 Several neurological manifestations have also been correlated with aPL, including chorea, myelitis, seizures, migraine, and cognitive impairment.17 Livedo reticularis can be the presenting clinical manifestation in approximately 40% of APS patients;19 this is a typical pattern of the skin, either mottled or reticular, with a colour ranging from reddish-blue to purple, and localised to the trunk, arms, or legs. The ‘regular livedo reticularis’ consists of regular unbroken circles, whereas the ‘livedo racemosa’ consists of irregular broken circles, and is also more generalised and irregularly distributed than the livedo reticularis.16,19 The rare Sneddon’s syndrome is the association of livedo, either reticularis or racemosa, and cerebrovascular events.20 The relevance of extra-criteria manifestations is still debated.
Although they are not currently included in the APS classification criteria, a report from the Antiphospholipid Antibodies Task Force on Clinical Manifestations suggested there is moderate evidence to support the inclusion of APS-nephropathy, valvular heart lesions, and livedo reticularis.18
Finally, on rare occasions a catastrophic variant of APS (CAPS), also known as Asherson’s syndrome,21 can develop. The prevalence of CAPS is <1.0% of all patients with APS; it is a potentially life-threatening condition with very high mortality rates (40–50%).22 CAPS is characterised by the rapid development of extensive microvascular thrombosis, leading to multiorgan failure. Any organ can be affected, but CAPS typically involves the kidneys, lungs, and central nervous system. Precipitating factors have been recognised in 65% of cases, most commonly infections, surgery, malignancies, and hormonal stimuli, such as oral contraceptives or pregnancy.22 The classification criteria for CAPS were established in 2002, and were defined as thrombosis in three or more organs, simultaneous development or development within a week, histopathological confirmation of small vessel occlusion, and laboratory confirmation of aPL (LAC and/or aCL, usually in high titre).23 The diagnosis of ‘definite CAPS’ requires all four criteria, while ‘probable CAPS’ is diagnosed when the four criteria are not completely fulfilled or with different combinations of three criteria. Particular laboratory findings reported in the international CAPS Registry24 include thrombocytopenia (67%), haemolytic anaemia (37%), schistocytes (22%), thrombotic microangiopathy (14%, defined as the association of thrombocytopenia, haemolysis, and schistocytes), and disseminated intravascular coagulation (11%, defined as the association of thrombocytopenia, increased D-dimer, and prothrombin time).
Diagnosis
Diagnostic Criteria
The diagnosis of APS is currently based on Sydney criteria:16 at least one clinical criterion (vascular thrombosis or pregnancy morbidity) and one laboratory criterion (LAC, aCL, or aβ2-GPI) should be met (Table 1). Vascular thrombosis can involve arteries, veins, or small vessels in any organ, and should be confirmed by appropriate imaging or histopathology. If histopathological confirmation is sought, thrombosis must be present without any sign of inflammation of the vascular wall. Superficial vein thrombosis is not included in the clinical criteria. Recurrent first trimester miscarriages are defined as ≥3 unexplained consecutive abortions before the 10th week of gestation, after having excluded maternal anatomic or hormonal abnormalities and maternal and paternal chromosomal abnormalities.16

Table 1: Classification criteria for the antiphospholipid syndrome (Sydney criteria).
Laboratory Tests
The presence of aPL should be confirmed by specific laboratory tests, for which the timing is crucial. Some groups argue that these tests should not be performed during the acute phase (the first 12 weeks) after a thrombotic event,16 to avoid false-positive results. However, these tests could give an indication of the actual diagnosis when it matters, such as in those patients with the catastrophic type of APS. In any case, a positive laboratory test should always be confirmed at least 12 weeks apart, to exclude the transient presence of aPL, which is common after infectious diseases.16,25,26 The diagnosis of APS should not be made if the positive laboratory test occurs >5 years after the clinical event.16 Finally, the LAC tests are best postponed until after discontinuation of anticoagulant treatment, to avoid interference in the prolongation of the basal clotting time.16 Practical suggestions have been reported: delay for 1–2 weeks after discontinuation of vitamin K antagonists (VKA) or when the international normalised ratio (INR) is <1.5; if bridging with low molecular weight heparin (LMWH), delay for at least 12 hours after the last dose; wait until after discontinuation of the direct oral anticoagulants (DOAC).27 In those patients who are treated with a DOAC, there is another way to remove the anticoagulant effect and allow testing, involving the addition of DOAC-Stop® to the plasma sample.28
The LAC should be tested according to the guidelines of the Subcommittee on Lupus Anticoagulant/Antiphospholipid Antibodies of the International Society on Thrombosis and Haemostasis.25,26 The tests should be expressed as a ratio between the coagulation time of the patient plasma and normal pooled plasma, and a multistep procedure of screening, mixing, and confirmation tests is usually recommended. Since no single test has enough sensitivity to account for antibodies’ heterogeneity, two phospholipid dependent clotting assays should be performed as screening tests to exclude the presence of LAC.16,26 The Dilute Russell viper venom time is the first test of choice, because of its specificity for clinically significant antibodies. The second test should be a sensitive activated thromboplastin time, using silica as an activator, and low phospholipid concentration to emphasise the effect of the LAC by competition for the limited phospholipid-binding sites.29 A positive screening test shows prolongation of the clotting time.26 The mixing test is performed by adding normal pooled plasma to patient plasma with a 1:1 ratio, to differentiate among the possible causes of prolonged clotting time. While coagulation factor deficiencies are corrected by the mixing test, the presence of coagulation inhibitors (such as LAC) still result in a prolongation of the clotting time.26 Finally, a confirmatory test is performed by increasing the concentration of phospholipid in the screening test, to overwhelm any aPL and demonstrate phospholipid dependence.29 A positive confirmatory test shows normal clotting time.
The recent development of integrated tests, which can perform screening and confirmation tests in parallel just by varying the concentration of phospholipid, has reduced the number of mixing tests.26 However, the role of the mixing test is still debated; while some authors acknowledge that it can introduce a dilution factor and generate false-negative results if the LAC is weak,30 others argue that the mixing test still has a role when the other test results are borderline29 or that skipping the mixing test might generate both false-negative and false-positive results.31
The aCL and aβ2-GPI are usually detected by enzyme-linked immunosorbent assays, but recently automated solid phase assays have also been developed.25,32 Only IgG or IgM isotypes at high titre are considered positive, defined as antibodies levels above the 99th percentile of a cut-off established locally on a population of healthy volunteers.25,32 IgG showed a strong association with the risk of thrombosis,32 while IgM showed possible false-positive results in the presence of cryoglobulins and rheumatoid factor, especially when at low titre.16
Guidelines recommend that all three tests (LAC, aCL, and aβ2-GPI) should be performed on the same sample to characterise the patient antibodies profile.25 The risk of clinical events increases in parallel with the number of positive tests and is especially high when all three tests are concomitantly positive, known as triple positivity. Furthermore, a recent study showed that 98% of patients with triple positivity and 84% with double positivity were confirmed at the 3-month follow-up, versus 40% with single positivity.33
Problems in APS diagnosis might arise from technical difficulties. Despite the efforts to standardise the laboratory diagnosis of aPL, the inter-laboratory variability in the detection of the LAC remains high.34 Furthermore, kits for the detection of autoantibodies, especially aCL, produced by different manufacturers may provide different results, even when performed in the same laboratory.35
Non-Criteria Antiphospholipid Antibodies
Several other autoantibodies not included in the laboratory criteria have been recently identified in APS patients. They include IgA aPL isotypes (IgA aCL and IgA aβ2-GPI), antibodies against prothrombin (aPT) or phosphatidylserine/prothrombin complex (aPS/PT), and antibodies against the domain 1 of β2-GPI.17 IgA isotypes could contribute to the identification of APS patients, but they are currently not considered a diagnostic marker for APS, the reason being that they often coexist with the IgG and IgM isotypes. Anti-prothrombin antibodies were recently reported as a risk factor for thrombotic events, especially aPS/PT.36 Autoantibodies directed only against epitopes in the domain 1 of β2-GPI were more frequently detected in patients with triple-positivity and they were associated with a history of thrombosis.37 The practical relevance of these non-criteria antiphospholipid antibodies is currently debated. However, there is recent evidence that they could be involved in APS pathogenesis and explain some of the seronegative APS.17
Patient Selection
Asymptomatic patients should not be routinely screened for aPL to avoid incidental findings of false-positive results, due to the poor specificity of these assays.26,32 The appropriateness of searching for aPL is high in young (<50 years of age) patients with unprovoked VTE or unexplained ATE, unusual site VTE, thrombosis or pregnancy complications associated with autoimmune diseases, or late pregnancy loss.26 The appropriateness is moderate in young patients with provoked VTE, recurrent spontaneous early pregnancy loss, or unexplained prolonged activated thromboplastin time, and is low in elderly patients with VTE or ATE.26
Prognosis
APS carries significant morbidity and mortality. Among the patients included in the Euro-Phospholipid project, mortality rates were 5.3% in the initial 5-year follow-up and 4.0% in the subsequent 5-year follow-up.38 Thrombotic events (such as myocardial infarction, stroke, and pulmonary embolism) were the most common causes of death (36.5%), followed by sepsis (26.9%), malignancies (13.9%), and haemorrhages (10.7%). Furthermore, the most recent follow-up showed that, despite antithrombotic treatment, 24.8% of patients developed thrombosis and 8.3% had obstetric complications.38
Antibody profile is the major risk stratification tool. Patients with a single isolated positivity are at low risk of clinical manifestations of APS,27 whereas patients with triple positivity showed the strongest association with thrombotic or obstetric events. In a cohort of 160 triple positive APS patients with a mean follow-up of 6 years, ATE or VTE occurred in approximately 34.0% of patients: 36 of 123 (29.3%) anticoagulated patients and 19 of 37 (51.4%) non-anticoagulated patients.39 Among APS patients who suspended anticoagulant treatment for different reasons, 43.3% had recurrent thrombosis during a median follow-up of 4.3 years and triple positivity was a strong predictive factor for relapse.40 Despite appropriate treatment, in triple-positive women, the likelihood of a live-birth neonate is only 30%, compared to approximately 80% in those with single LAC positivity.41
Furthermore, triple positivity in asymptomatic aPL carriers is associated with a considerable risk of developing a first thrombotic event.42,43 It has been estimated that the annual rate of a first vascular event is 0.40% in normal subjects, 1.36% in single positivity aPL carriers, and 5.30% in triple positivity aPL carriers.42 Another study reported that the annual rate of a first vascular event was 0.65% in single positivity aPL carriers and 1.27% in double or triple positivity aPL carriers.43 In both studies, approximately a third of patients were receiving prophylactic low-dose aspirin, which was not associated with reduced risk of arterial or venous thrombotic events.42,43
A global APS score (GAPSS) was recently developed to predict the clinical manifestations of APS: thrombosis and pregnancy loss.44 The GASPSS includes a number of variables: aCL (5 points), LAC (4 points), aβ2-GPI (4 points), anti-prothrombin/phosphatidylserine complex (3 points), hyperlipidaemia (3 points), and arterial hypertension (1 point). The adjusted GAPSS (aGAPSS) is a simplified version, which excludes the antibodies against prothrombin/phosphatidylserine, since they are not routinely tested and not included in the classification criteria for APS.45 The GAPSS and aGAPSS have been validated in different populations, including patients with SLE or other systemic autoimmune diseases (to predict the first manifestations of APS) and in patients with primary or secondary APS (to predict recurrent events).46
Treatment
Anticoagulant Treatment
In APS patients presenting with VTE, the standard initial treatment involves unfractionated heparin (UFH) or LMWH, followed by VKA with INR target range 2.0–3.0.30 This recommendation is based on the results of two randomised controlled trials (RCT) (Table 2) showing that a higher INR target range was not associated with a further reduction of recurrent thrombosis.47,48 Considering the high risk of thrombosis recurrence after discontinuation,49 anticoagulant treatment duration should be long-term for APS patients with unprovoked VTE, while the benefit of extended anticoagulation in APS patients with VTE provoked by a transient risk factor is still debated.50
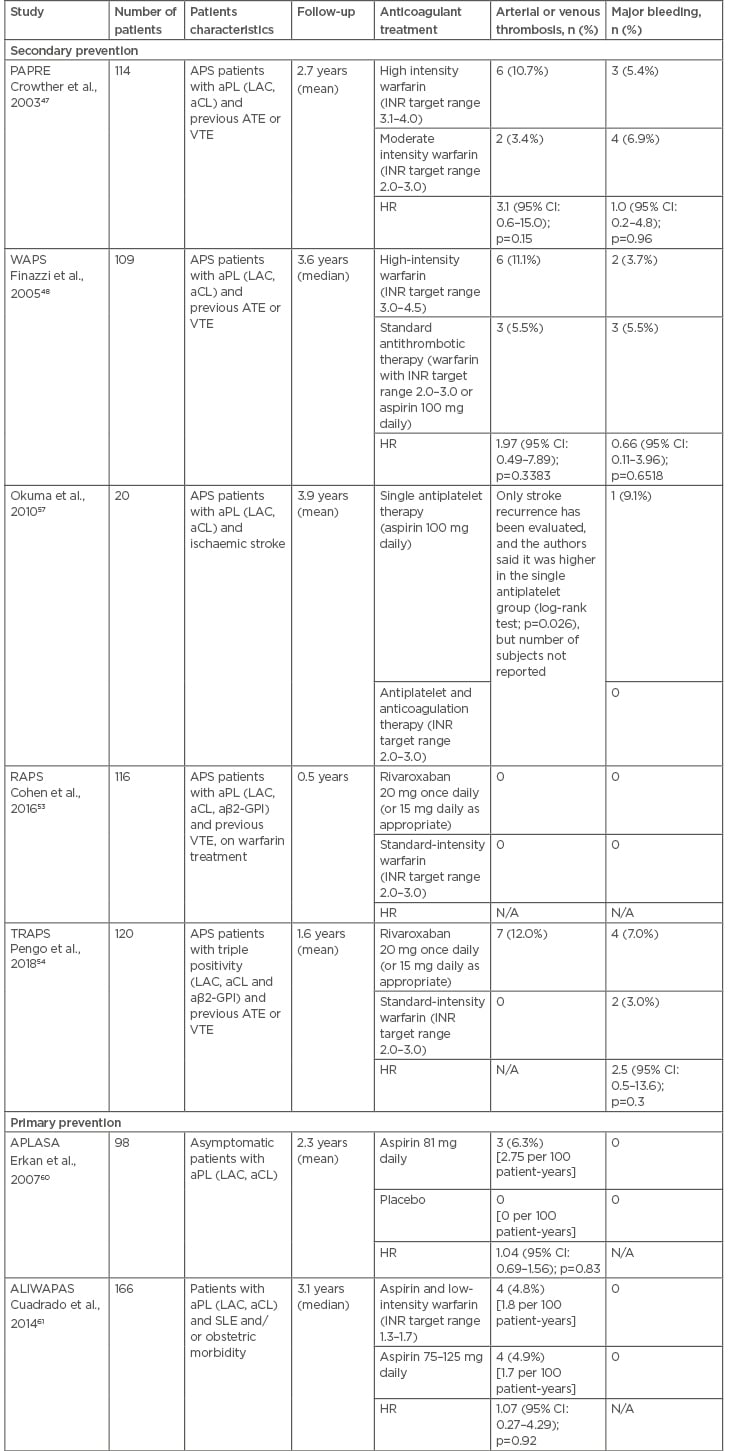
Table 2: Randomised controlled trials evaluating the antithrombotic treatment in patients with antiphospholipid antibodies.
aβ2-GPI: anti-β2 glycoprotein-I; aCL: anticardiolipin antibodies; aPL: antiphospholipid antibodies; APLASA: antiphospholipid antibody acetylsalicylic acid; APS: antiphosphoplipid syndrome; ATE: arterial thrombotic events; CI: confidence interval; HR: hazard ratio; INR: international normalised ratio; LAC: lupus anticoagulant; N/A: not applicable; PAPRE: patients with antiphospholipid antibodies prevent recurrent events; RAPS: rivaroxaban in antiphospholipid syndrome; SLE: systemic lupus erythematous; VTE: venous thromboembolism; WAPS: warfarin in the antiphospholipid syndrome.
Monitoring VKA can be difficult in APS patients. Since certain commercial thromboplastins used to measure the prothrombin time are more sensitive to LAC than others and can cause artifactual prolongation of the INR, and thus subtherapeutic VKA dose,51 lupus insensitive reagents should ideally be used. The LAC interference can also be seen with the use of some point-of-care INR devices; caution is therefore recommended.30,51
Recurrent thrombosis during VKA treatment at therapeutic INR (warfarin failure) is a known complication of APS52 and can lead to different management strategies, such as increasing the INR target range, shifting to LMWH, or adding low-dose aspirin.50
There are two published RCT evaluating the use of rivaroxaban compared to warfarin in APS patients (Table 2). The RAPS trial used a surrogate endpoint (the change in the endogenous thrombin potential from randomisation to Day 42) and showed no clinical events in either groups during the 6-month follow-up.53 The TRAPS trial enrolled only APS patients with triple positivity and was prematurely interrupted due to an excess of arterial thrombotic complications in the rivaroxaban arm.54 A RCT evaluating apixaban is ongoing.55
The treatment of APS patients presenting with ATE is less defined, since few patients with ATE were enrolled in the RCT evaluating the VKA.47,48 Possible therapeutic options include aspirin alone (e.g., in elderly patients presenting with stroke), VKA at standard INR target range or high-intensity warfarin (INR target range 3–4),50 and the combination of VKA and low-dose aspirin (e.g., after failure of single antithrombotic therapy).56,57 It is also important to act on risk factors for ATE, such as hypertension, hyperlipidaemia, obesity, and smoking.
In women with obstetrical APS, the combination of prophylactic-dose LMWH (or prophylactic/intermediate-dose UFH) and low-dose aspirin is usually prescribed to prevent pregnancy complications,58 based on the evidence that this association may halve the risk of pregnancy loss.59 Heparin should be continued for 6 weeks after birth, because of the high thrombotic risk during the puerperium.50 Close monitoring of the fetus and the mother during pregnancy is also suggested, to identify placental insufficiency or fetal distress.5
The need for primary thromboprophylaxis in asymptomatic aPL carriers without any previous thrombotic event is debated. Primary prevention RCT (Table 2) showed scarce benefit of low-dose aspirin or aspirin and warfarin, considering the low annual incidence rate of thrombosis in this population.60,61 The guidelines of the British Committee for Standards in Haematology discourage the use of primary thromboprophylaxis in individuals with incidentally discovered aPL.30 Vice versa, the consensus document elaborated by an international Task Force at the 13th International Congress on aPL recommends low-dose aspirin for SLE patients with aPL and suggests the same thromboprophylaxis for non-SLE individuals with high-risk aPL profile (LAC, triple positivity, or aCL at medium-high titre).62 It has been estimated that the annual risk of a first thrombotic events is <1% in subjects with aPL without any other risk factors, compared to 5% in patients with a high-risk aPL profile associated with systemic autoimmune diseases.50
Other Treatments
Additional treatment strategies are used to address the clinical manifestations of APS. Hydroxychloroquine has anti-inflammatory and anti-thrombotic properties and is recommended by recent consensus guidelines as primary thromboprophylaxis in patients with aPL, SLE, and no contraindications, where it was shown not only to protect against thrombosis, but also to increase survival.62 Furthermore, hydroxychloroquine may have a role as adjuvant therapy in APS with recurrent thrombosis despite adequate anticoagulant treatment.50,62
Statins have pleiotropic effects, including anti-inflammatory and anti-thrombotic properties. Two recent studies showed that fluvastatin can reduce pro-inflammatory and pro-thrombotic markers in aPL patients, while pravastatin can improve pregnancy outcomes in women with obstetrical APS.6
Rituximab is a monoclonal antibody against CD20 located mainly on B-lymphocytes. There is recent evidence that rituximab may be effective in controlling some non-criteria manifestations of APS (such as thrombocytopenia, haemolytic anaemia, and skin ulcers) and can also be an option for refractory CAPS.50,63 Another monoclonal antibody, eculizumab, a C5 complement inhibitor, can be effective in refractory CAPS, blocking the widespread complement activation, and preventing recurrent APS post-kidney transplantation in patients with aPL-related nephropathy.50
Catastrophic Antiphospholipid Syndrome
Treatment of CAPS should aim at the control of any precipitating factor, as well as the prevention and treatment of thrombosis. In a proposed treatment algorithm for CAPS, prompt use of anticoagulation (usually intravenous UFH) and high dose corticosteroids represented the first-line option, with the addition of intravenous immunoglobulin and/or plasma exchange (to remove pathogenic aPL and the excess of cytokines) in life-threatening conditions.23 In the CAPS registry, the combination of anticoagulant, steroids, and plasma exchange obtained a recovery rate of 77.8%.64 Rituximab and eculizumab are second-line options in refractory CAPS, although they are only supported by a few case-reports.50
Conclusion
APS is a rare autoimmune disease characterised by significant morbidity and mortality. The diagnosis of APS needs the simultaneous presence of clinical and laboratory criteria. Laboratory tests should be performed according to international guidelines, which are periodically updated. The treatment of APS patients presenting with VTE, ATE, or obstetrical APS may differ, but anticoagulation usually plays a substantial role. New emerging treatments may provide additional strategies to address the clinical manifestations of APS in the future.