Abstract
This article relates the current pathophysiologic and radiologic findings to the fundamental idea of acute lung epithelial infection, alveolar inflammation causing leak into the interstitial space, and subsequent secondary or concurrent endothelial infection and dysfunction. Understanding the mechanisms and timings of alveolar damage can better inform the types of ventilatory support required and timing of targeted pharmacotherapies.
INTRODUCTION
Coronavirus disease (COVID-19) is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2);1 as of September 2020, there were >33 million cases and almost 1 million deaths worldwide.2 An unknown number of people are asymptomatic carriers of SARS-CoV-2 but symptoms in the early phase of viral replication include fatigue, anosmia, fever, sore throat, and cough. Around 85–95% of people make an uncomplicated recovery within 10 days; however, some individuals develop worsening cough, dyspnoea, and persistent (higher) fever, some of whom require hospital admission with hypoxia and interstitial pneumonia.3 This more severe illness is from viral infection within the lung tissue and the associated immune dysfunction, with loss of regulation of the inflammatory cascade leading to further lung damage, worsening hypoxia, and other organ dysfunction.
FUNDAMENTAL PATHOPHYSIOLOGY
Two possible inflammation mechanisms (not mutually exclusive) can result in fluid accumulation in the lung alveoli: 1) direct viral and immune-mediated damage to the epithelium, affecting alveolar movement and reducing pulmonary fluid clearance; 2) a secondary or parallel injury to the endothelium or its basement membrane that results in capillaritis with further leak of fluid from the plasma into the interstitial space and thereon into the air spaces.
IMPACT OF EPITHELIAL DAMAGE
SARS-CoV-2 targets and damages pulmonary epithelium,4 particularly ciliated airway epithelial cells and Type 2 pneumocytes through the abundant angiotensin-converting enzyme 2 receptors in the lung. The virus relies on cell membrane serine protease TMPRSS2 cleaving and subsequently activating S-proteins and furine protease activity.5 Hydrostatic and osmotic forces determine fluid movement from the vascular space to the interstitium through the extracellular matrix and the endothelial glycocalyx layer. Once the normally tight alveolar epithelial barrier is breached, alveolar oedema ensues. The epithelium also performs an important function in reabsorption of fluid through the asymmetric distribution of sodium channels and pumps throughout the alveoli and small airways. Sodium from the alveolar fluid passively enters the apical part of epithelial cells via the sodium channel and is pumped out of the cell via a sodium–potassium pump on the basal membrane. This shift of sodium causes an osmotic gradient leading to movement of water out of the alveoli, avoiding or resolving oedema in the alveolar air space (Figure 1).6 Inflammatory damage that increases permeability of the epithelial barrier or a reduction in the number of epithelial cells, channels, and/or pumps would therefore impair an important clearance mechanism that helps avoid or resolve pulmonary oedema. Hypoxia itself is extremely proinflammatory and can further exacerbate tissue damage through cytokine release in a self-perpetuating and rapidly escalating autoimmune cycle of harm. Hypoxia further attenuates the basal pumps’ highly energy-dependent sodium-potassium pump clearance mechanism.
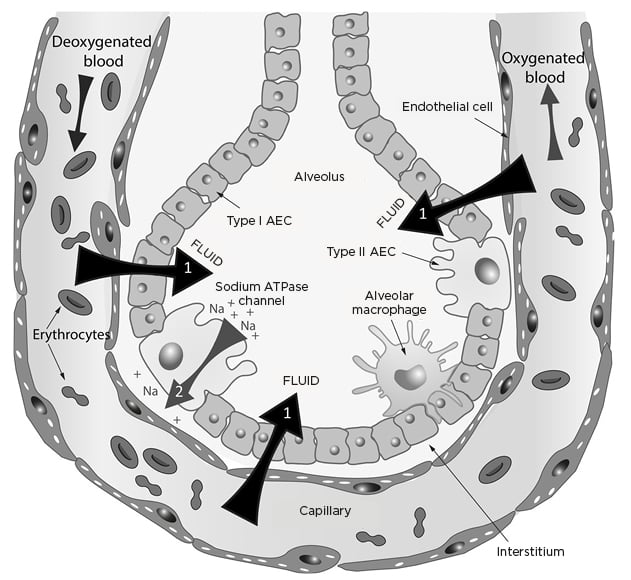
Figure 1: Fluid movements across the alveolar–capillary barrier (black and grey arrows).
AEC: alveolar epithelial cells; Na: sodium.
POTENTIAL VENTILATION OR VENTILATOR DAMAGE TO THE PULMONARY ENDOTHELIUM OR EPITHELIUM
Normal breathing at tidal volume can further stretch damaged and friable epithelium or endothelium and increase the permeability further. Excessive movements of damaged alveolar units through mechanical stress or sheer stress exacerbate extravasation of fluid, worsening hypoxia. In this regard, the authors propose that ventilatory support strategies should allow enough alveolar capillary oxygenation with a minimal degree of alveolar movement. Reducing physical movement of a leaky epithelium would reduce leak, attenuate further hypoxia, and buy more time for the body to clear the virus or for the antivirals to take effect, and for timely, targeted immunosuppression to reduce bystander cellular damage.
EVIDENCE FROM PULMONARY IMAGING
CT chest scans report ground-glass opacities, often together with areas of consolidation and lung interlobular septal thickening, predominantly in subpleural and middle/lower lung fields.7-10 Plain chest radiographs show multiple patchy shadows,7,8,11 and ultrasounds report B-lines ranging “across a continuum from mild alveolar interstitial pattern, severe bilateral infiltration pattern to lung consolidation”.12
Serial CT scans show early subpleural bilateral patchy focal infiltrates, which coalesce into large confluent patchy areas, becoming geographically connected in a ‘crazy-paving’ pattern and ultimately progressing to diffuse lung consolidation.13,14 These sequential images represent progressive areas of alveolar epithelial damage, consistent with a spreading infection and local associated immune reaction. This patchy distribution supports an aerogenous route of pulmonary infection.15 CT scans usually correlate with clinical severity,7 but unlike traditional models of acute respiratory distress syndrome (ARDS) there can be severe radiological changes in relatively well and even asymptomatic people. Conversely, some symptomatic patients may present with a normal CT, especially in the early phase.16
Imaging in acute COVID-19 is entirely consistent with patchy progressive infection and distal alveolar inflammatory leak.12 Pleural effusions are rare in COVID-19 because the leak into the third space is not due to hydrostatic back-pressure, but due to small multifocal areas of patchy alveolar epithelial and endothelial damage.7,9
EVIDENCE FROM GROSS LUNG PATHOLOGY
Gross thoracic changes include uneven mottled red to blue-red colour, increased lung weight, oedema, severe congestion, pleuritis, consolidation thrombi, and often secondary bacterial infection.17,18
EVIDENCE FROM LUNG HISTOPATHOLOGY
Antemortem histopathological changes were seen coincidentally in samples of lung tissue taken for other reasons. Findings included alveolar oedema, fibrinous exudate, inspissated protein, haemorrhage, mononuclear inflammation, and multinucleated giant cell formation.18,19 Two patients developed severe COVID-19, and one of these patients died.20
Postmortem histopathological changes included desquamation of the alveolar epithelium, reactive Type 2 pneumocyte hyperplasia, intra-alveolar exudates with organising fibrin, hyaline membrane formation, haemorrhage, excess lymphocytes, neutrophils, alveolar megakaryocytes, thrombosis of small and midsized pulmonary arteries, and some interstitial fibrosis.14,21-23 From the autopsies of 21 individuals who died as a result of COVID-19, the primary cause of death was reported as respiratory failure, with exudative diffuse alveolar damage, and with severe capillary congestion and microthrombi in alveolar capillaries. Pulmonary embolism (n=4), alveolar haemorrhage (n=3), vasculitis (n=1), and superimposed bronchopneumonia (n=10) were also reported.18
The inflammation is predominantly lymphocytic with large atypical pneumocytes,11 probably caused by viral cytopathic changes.11,18,24 Macrophages and monocytes are increased in alveolar spaces, and virus particles or antigens are detected in the epithelial cells, Type 1 and 2 pneumocytes, and macrophages by electron microscopy, immunohistochemistry, and in situ hybridisation.21
The primary damage occurs mainly to the alveolar epithelial cells. Initially, some research proposed that the lack of direct damage to endothelial cells may partly explain why other organs (heart: 7–9%; kidney: 3–15%) are less affected than the lungs.25-28 However, COVID-19 clearly incorporates other organ dysfunction and much more so than in other coronaviruses (severe acute respiratory syndrome and Middle East respiratory syndrome) and influenza.29
Endothelial cell viral infection, inflammatory cell infiltration, apoptosis of endothelial and mononuclear cells with widespread thrombosis,30 microangiopathy, marked increase in fibrinous microthrombi, and both intussusceptive and conventional sprouting angiogenesis31 have been reported. In one systematic pulmonary dissection study, all patients exhibited thrombosis of small or midsized pulmonary arteries.22 Here, the authors postulate that direct infection of endothelial cells does occur and their close proximity to the epithelium in the lung is why the lung is affected most by local vascular dysregulation,22,30,32 leading to further hypoxia without loss of lung compliance in the early phase.
EVIDENCE FROM PATHOPHYSIOLOGY
Intubated Patients
Standard ARDS Berlin criteria require an onset within 7 days of a clinical insult or respiratory symptoms;33 however, in COVID-19, respiratory failure occurs typically 10–15 days after onset of symptoms.25-28 Early cohorts had worse outcomes with over-inflation34,35 and higher levels of barotrauma compared to ventilated patients with standard ARDS.35 Current COVID-19 ventilator guidelines consistently recommend lower pressures, probably to reduce alveolar stretch.36,37
Unlike most people with ARDS, many patients with COVID-19 receiving ventilation (especially early on) have compliant lungs despite profound oxygenation failure.38,39 Some studies have defined separate phenotypes in COVID-19 pneumonitis according to lung compliance,40 but whether these are truly distinct or sequential stages of the same disease is in debate. Relatively high compliance is associated with high shunt fractions (50%), rarely seen in other forms of ARDS.40 Again, this supports the idea that disrupted vasoregulation is an important contributor to poor oxygenation, at least in the early stages of COVID-19 lung injury. A major problem is the failure of the normal hypoxia-induced pulmonary vasoconstriction,41 which depends on good endothelial function, leading to hyperperfusion of ‘gasless tissue’. CT scans report areas of dilated vessels in parts of the lung that are unlikely to be well ventilated.10,41 The improvement of oxygenation with positive end-expiratory pressure and prone positioning may be due to haemodynamic adjustment through local fluid shifts and better alveolar recruitment.39
The origin of the ‘preserved’ compliance is unknown; while it is probably beneficial for ventilation, it may have other detrimental effects. The lung possesses a highly efficient mechanism to minimise interstitial fluid volume. The pulmonary capillary basement membrane normally has low permeability to water due to the macromolecular organisation of heparan sulfate proteoglycans. Furthermore, any increase in extravascular fluid is rapidly cleared because of the high elastance (poor compliance) of the extracellular matrix consisting of a second group of matrix chondroitin sulfate proteoglycans. Hypoxia causes fragmentation of both sets of proteoglycans, resulting in decreased elastance (increased compliance) and leading to more alveolar movement and potential oedema.42 Applying high levels of positive end-expiratory pressure may accentuate underlying alveolar and microvascular injury and contribute to a worse outcome.43
The authors suggest that the respiratory distress seen in COVID-19 starts with a primary epithelial failure, followed by a secondary or concurrent endothelial vascular dysfunction. Therefore, ventilation should be guided by daily changes in radiology and estimates of capillary wedge pressure and lung water. Cardiac dysfunction from viral-induced myocarditis and/or hypoxaemia, worsening any pre-existing cardiac conditions, may also complicate vascular filling. Various fluid balance strategies have been tried in typical ARDS, based on estimated pulmonary capillary wedge pressure and extravascular lung water, but the unusual pulmonary haemodynamics seen in COVID-19 are a further complicating factor in the cardiac–pulmonary-specific interactions in intubated patients.
The wide variations in mortality of intubated patients with COVID-1926,38,39 could be due to fixed ventilatory strategies and basing pressure or target volumes on traditional ARDS models. The authors argue that the traditional (Berlin) definitions of ARDS do not align with COVID-19 respiratory failure in terms of diagnosis or, more importantly, ventilator management of lower pressures and daily changes according to variable haemodynamics and hypoxaemia. Extracorporeal membrane oxygenation (ECMO) is increasingly used for adult patients with potentially reversible respiratory failure refractory to conventional management. ECMO beds have been increased worldwide in response to the pandemic and some countries have issued guidelines to assist in identifying eligible patients and management.44 However, most clinicians in the ECMO service agree that the current evidence base does not allow strict criteria to determine optimal benefit and therefore encourage early referral. In reality, the experience (of the authors) has been that decisions are often made according to ECMO availability.
Awake Patients
Hypoxic patients with COVID-19 are commonly seen taking rapid and shallow breaths.45 Increased pulmonary interstitial pressure or volume activates pulmonary afferent C-fibre receptors, which are known to initiate a reflex that includes rapid shallow breathing and dyspnoea.46 These receptors are stimulated by increases in pulmonary interstitial pressure or volume (as occurs in pulmonary oedema). The combination of hypoxia, hypocapnia, tachypnoea and pulmonary oedema as seen in altitude sickness,47 traumatic ‘blast lung’,48 and chemical lung injury49 are explained by the increased minute ventilation in response to the hypoxia; there is a disproportionate elimination of CO2 because its exchange is not impaired by pulmonary oedema as much as oxygen.
Awake patients with COVID-19 tolerate extreme hypoxia surprisingly well. Many patients converse freely despite peripheral oxygen saturations of 80–92% but have respiratory rates of 30–38 breaths per minute, suggesting that rapid, shallow breathing is a natural response. Deeper inspiration causes dyspnoea and often back or chest pain in individuals with radiological COVID-19 pneumonitis (personal observation from Lewis KE).
However, if patients experience worsening hypoxia and start taking strong, spontaneous inspiratory efforts, this can physically stretch an already damaged alveolar epithelium and increase tissue stresses (akin to patient self-inflicted lung injury).50 Coexistent endothelial dysfunction with ventilation/perfusion mismatch leads to rapidly worsening hypoxaemia, more inflammation, more leak, and more hypoxia.
However, one notable difference is that dyspnoea is much less pronounced in COVID-19. Fifteen percent of patients with mild-case COVID-19 had shortness of breath, and only 38% of severe cases in China27 and 17% of hospitalised cases in New York, USA, with tachypnoea51 complained of breathlessness. Whether this is of any significance in terms of respiratory control mechanisms remains to be seen.
Patients experiencing mild COVID-19 pneumonitis often have rapid, shallow breathing at rest, with normal arterial oxygen saturation. Frequently in these patients, mild exercise precipitates peripheral oxygen desaturation, consistent with an initial small reduction in pulmonary capillary gas transfer, which is sufficient to activate the pulmonary afferent C-fibre receptors and alter the breathing pattern.46 In normal lungs at rest, however, because the blood becomes fully saturated with oxygen by the time it has travelled approximately one-third the length of the pulmonary capillary, there is sufficient reserve to allow nearly complete saturation when the patient is at rest.52 However, with mild exercise the increased cardiac output reduces pulmonary blood transit time, so that pulmonary gas transfer now becomes diffusion-limited with consequent arterial desaturation. During recovery and rehabilitation, patients still profoundly desaturate, suggesting that it takes some time for the pulmonary capillary diffusion barriers to completely resolve.
SUPPORTING EVIDENCE FROM CONTINUOUS POSITIVE AIRWAY PRESSURE
Despite early experience with invasive ventilation, many healthcare professionals are moving towards noninvasive ventilation strategies for hypoxic COVID-19.53 Mild-to-moderate oxygenation failure in COVID-19 responds to high-flow nasal oxygen (HFNO) systems or continuous positive airway pressure (CPAP).3 These treatments allow the patient to eat, drink, communicate, and self-position. By reducing the work of breathing and inspiratory effort, it can reduce mechanical sheer stress, ventilation-induced lung injury, tracheostomy, ventilation-associated pneumonia, and difficulty in weaning. CPAP may delay or avoid the need for mechanical ventilation in patients with COVID-19.27,53-55 When intensive treatment unit resources are scarce, inappropriate use of invasive ventilation may stop access to life-saving treatment to those that need it. CPAP is part of the UK recommendations for the management of COVID-19.41,56 It is effective in the treatment of chemical lung injury with pulmonary oedema, reduced gas transfer, and high shunt fraction.57
These pathophysiological mechanisms explain how a ventilatory support strategy using CPAP and HFNO maintains both a physical pressure gradient and a partial-pressure gradient to allow continuous oxygen diffusion down the airways and across gently splinted-open alveoli that do not need to move much. This also allows rapid, small breaths to minimise movement of damaged epithelial and endothelial interfaces and supports fatigued muscles. Combining CPAP with HFNO allows maximum diffusion at the alveolar–capillary interface with minimum alveolar movement (Figure 2).
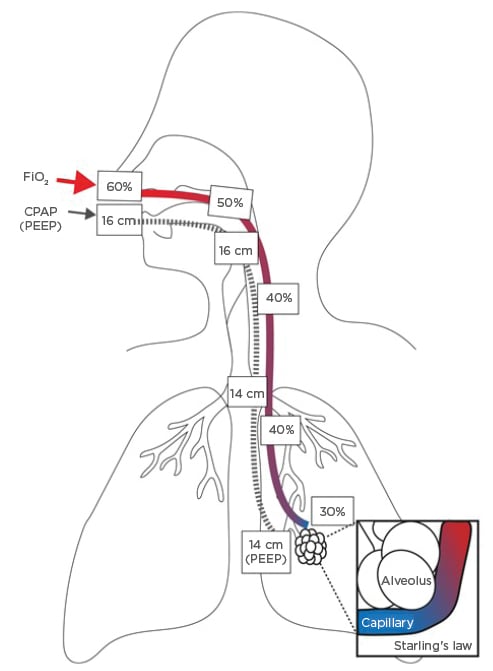
Figure 2: Combined benefits of airway pressure and oxygen.
CPAP: continuous positive airway pressure; FiO2: fraction of inspired oxygen; PEEP: positive end-expiratory pressure.
ADDITIONAL IMPORTANT TREATMENTS
Adequate fluid replacement is important as patients are prone to dehydration through increased insensible losses (fever, tachypnoea, occasionally diarrhoea) and reduced oral intake (nausea, CPAP masks).
Prone positioning can help oxygenation in some patients (while awake, on CPAP and ventilators). Presumably, prone positioning helps recruit more or different alveoli and therefore aligns with the hypothesis of localised inflammation and alveolar leak, rather than alveolar oedema from heart failure or increased hydrostatic pressure, which often worsen when lying supine or prone.
Management of coexistent medical conditions, especially diabetes, cardiac dysfunction, pulmonary emboli, and superadded bacterial or fungal lung infections, is also crucial. Worsening symptoms (including new confusion), worsening hypoxia, higher ventilatory pressure support, new raised temperature, rising inflammatory markers (neutrophils and C-reactive protein), raised procalcitonin, and especially focal consolidation and fluid or cavities on chest X-rays or CT scans, should alert clinicians to coincidental/secondary infections. Bacterial and fungal infections are reported in at least 10–30% of postmortem lung specimens of people who died of COVID-19.18,21-23
SUPPORTING EVIDENCE FROM DRUG TREATMENTS
A detailed review of all current pharmacological treatments is beyond the scope of this article. However, the predominant success so far of immunosuppressant and immunomodulatory drugs fits with the idea of reducing primary inflammatory epithelial/endothelial damage. The UK multicentre RECOVERY trial showed dexamethasone reduced deaths in ventilated patients (rate ratio: 0.65; 95% confidence interval [CI]: 0.48–0.88; p=0.0003) and deaths in patients receiving oxygen (rate ratio: 0.80; 95% CI: 0.67–0.96; p=0.0021). There was no benefit among those patients not requiring respiratory support (rate ratio: 1.22; 95% CI: 0.86–1.75; p=0.14)58 presumably because there was no bystander immune-related damage at this point.
A meta-analysis showed that systemic steroids improve mortality in critically ill patients with COVID-19, likely due to cytokine storms and because steroids reduce bystander immune-related alveolar damage.59
Pathogenic T cells and inflammatory monocytes incite cytokine storms with large amounts of IL-6; therefore, monoclonal antibodies that target the IL-6 pathways may potentially curb these inflammatory storms. Tocilizumab, a monoclonal antibody against IL-6, appears to reduce mortality by approximately 24% and duration of organ support by around 1 week, in patients in intensive care units who need ventilatory support and have already received steroids (number needed to treat: 12).60 Tocilizumab may not prove to be so effective in patients less critically ill and who only require oxygen.61
Other inflammatory and immunomodulatory drugs blocking endothelial activation and injury62 or boosting pulmonary epithelial cell clearance of alveolar oedema (Figure 1) may also be useful.63 Other therapies such as the antiviral drugs remdesivir, lopinavir, hydroxychloroquine, and interferon have not been shown to reduce mortality or initiation of ventilation.64 Convalescent plasma and hyperimmune immunoglobulins are still being tested.
CLINICAL CASES
The following clinical cases illustrate the pathophysiology and response to CPAP treatments.
A 59-year-old male was brought to hospital by ambulance. He was mildly overweight and had controlled hypertension. On arrival, his peripheral capillary oxygen saturation (SpO2) was 67%, despite receiving oxygen 15 L/min via a nonrebreather mask. Arterial blood gases confirmed severe Type 1 respiratory failure (partial pressure of arterial oxygen: 5.3 kPa; partial pressure of CO2: 4.3 kPa). His quick sepsis-related organ failure score was 1 (blood pressure: 130/60 mmHg; respiratory rate: 38 breaths/min; Glasgow coma score: 15) but despite hypoxia he was able to speak on the phone. SpO2 improved to 80% and respiratory rate reduced to 24 breaths per minute with nasal CPAP (12 cmH2O pressure) with entrained oxygen at 15 L/min. Improvement in temperature and oxygenation occurred 5 hours after receiving 500 mg of intravenous methylprednisolone. CT confirmed bilateral extensive ground-glass shadowing and confluent infiltrates. A swab was positive for viral RNA of SARS-CoV-2. He was not intubated but continued CPAP with HFNO for 15 days with gradual weaning of both. He received six further doses of high-dose steroids (pre-RECOVERY results) and was discharged after 21 days.
Figure 3 displays transcripts from real-time videos of ward monitors from Patient 2 (53- year-old male, nonsmoker) and Patient 3 (58-year-old male, nonsmoker). Neither had any known lung or heart disease. Both patients were receiving 15 L/min of oxygen entrained through their CPAP machine (Trilogy Evo, Philips, Amsterdam, the Netherlands) set at 12–15 cmH2O pressure.
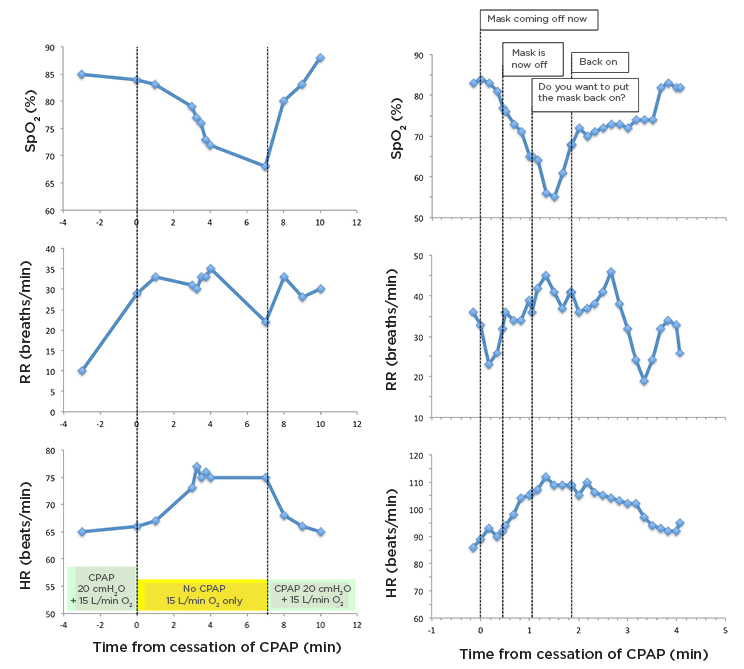
Figure 3: Changes in arterial oxygen saturation (skeletal muscle oxygen saturation, pulse oximetry), RR, and HR, on replacement of CPAP with high-flow oxygen during clinical care (to allow eating and drinking), and subsequent reintroduction of CPAP.
Comments on traces for Patient 3 were taken from audio recording coincident with cardiorespiratory monitoring.
CPAP: continuous positive airway pressure; HR: heart rate; RR: respiratory rate; SpO2: arterial oxygen saturation.
The graphs demonstrate abrupt falls in oxygen saturations and rises in pulse and respiratory rate, because the CPAP mask was removed to allow the patients to drink and take short breaks. Both were immediately switched to oxygen nasal cannulas at 15–20 L/min. Neither felt unduly breathless on removing their masks; however, Patient 3 reported feeling tired. Their SpO2 and pulse improved within 20 seconds of reapplying the positive pressure, suggesting that combining CPAP with HFNO is beneficial in people where oxygen alone is not sufficiently effective.
CONCLUSION
By relating underlying changes in epithelia, subsequent endothelial infection and dysfunction, and the pathophysiology of alveolar leak, the different disease stages and why certain combinations of treatments are most effective can begin to be understood. More importantly, the rapid increase in knowledge, based initially on observational physiology and now interventions, supports the concept of sequential epithelial and endothelial failure and explains differences in individuals over time and the variable response to treatments. Most importantly, understanding why and how pathophysiology changes so quickly allows for tailored interventions. Appropriate selection of early CPAP and timely immunosuppression should further improve outcomes. Certainly, one (treatment) size does not fit all, and one size will not fit one person over time.