
Abstract
Cholangiocarcinomas (CCA) are uncommon malignant tumours and are classified as intrahepatic, perihilar, or distal, depending on where they arise within the biliary epithelium. Surgery is still the only curative treatment, yet diagnosis is often made too late for this to be a viable option. For patients with locally advanced (unresectable), metastatic, or recurrent CCA, guidelines recommend palliative first-line chemotherapy with platinum compounds plus gemcitabine. However, the benefits are limited, with median overall survival being below 1 year, and there is an urgent need for novel, more effective treatment options.
Next-generation sequencing has revealed information about the genetic makeup of the CCA subtypes and CCA, especially intrahepatic, rank among tumours with the highest rate of potentially actionable gene alterations. A number of next-generation sequencing platforms are now commercially available, opening up the possibility for routine molecular testing at the time of diagnosis to allow a more personalised, targeted treatment approach. However, despite the availability of these platforms, barriers to their use remain, including issues with reimbursement in some countries.
Several clinical trials have been completed or are underway in CCA, investigating treatments directed against potentially actionable targets, such as FGFR2, IDH, NTRK, BRAF, and HER2. Some of these treatments are showing promising efficacy. Alongside, or before, initiating standard chemotherapy, efforts should be made to identify specific targets in all patients via molecular testing and treat eligible patients accordingly or enrol them in appropriate clinical trials.
INTRODUCTION
Cholangiocarcinomas (CCA) belong to the constellation of biliary tract cancers (BTC), along with gallbladder carcinomas and ampullary carcinomas. Among this heterogeneous group of uncommon tumours, CCA are notable for their high frequency of molecular alterations that are potentially amenable to therapeutic interventions. As such, CCA may be the ideal playground for a systematic policy of molecular testing. In this review, the authors underscore the need for early molecular testing in patients with advanced CCA as a result of the poor prognosis and limited therapeutic arsenal; the critical role of close communication between the molecular tumour board and the treating clinicians; co-operation in recommending appropriate novel tests, targeted therapies, or clinical trials based on the interpretation of individual patient’s data to fulfil the unmet needs in CCA; and the need for patient education, which should facilitate personalised decision-making, particularly when considering participation in appropriate clinical trials.
EPIDEMIOLOGY AND DIAGNOSIS
CCA are tumours of the biliary epithelium. They are categorised according to anatomical location into intrahepatic, perihilar, and distal. Intrahepatic CCA (iCCA) comprise 10–20% of all CCA1 and arise above the second-order bile ducts within the liver parenchyma. They can be further subdivided into large-duct type, resembling CCA that arise outside the liver, and smallduct type, which share pathological, aetiological, and imaging characteristics with hepatocellular carcinoma.2 Perihilar CCA (pCCA) involve the hilar region where the left and right bile ducts exit the liver and join to form the common hepatic duct; they make up 50% of all CCA.1 Distal CCA (dCCA), which account for 30–40% of all CCA,1 involve the common bile duct outside the liver.
CCA is the second most common hepatic cancer after hepatocellular carcinoma.3 In most countries it is rare, with an annual incidence of <6 cases per 100,000, but certain countries, such as Thailand and South Korea, have a particularly high incidence.4 Epidemiological studies suggest that rates of iCCA are increasing, particularly in Western countries; conversely, the incidence of both pCCA and dCCA appears to be declining.5 However, inconsistences in the classification of the subtypes indicate that these trends should be interpreted with caution.5 Worldwide, the average age at presentation is 50 years, although this is closer to 70 years in Western countries.6 CCA is slightly more common in males than females; data from the USA suggest a male:female ratio of 1.5:1.0.6
There are several risk factors for CCA, including biliary cysts and stones, primary sclerosing cholangitis, chronic liver diseases that lead to liver fibrosis (e.g., viral hepatitis), and congenital biliary tree abnormalities.6-8 In parts of Asia, CCA is often associated with liver fluke infestation and hepatolithiasis,6,7 but most cases arise in the absence of any known risk factor.7,9
Early diagnosis of CCA is challenging because most patients with early-stage disease are asymptomatic or have mild and/or nonspecific symptoms,10 particularly those with iCCA, in which jaundice is a late symptom. These patients may be identified incidentally with imaging studies or testing for liver enzyme abnormalities.
CURRENT TREATMENT OPTIONS FOR CHOLANGIOCARCINOMA
Patients with early-stage disease should undergo frontline surgical resection with curative intent; however, most patients are diagnosed too late for this to be feasible.4 Even after curative intent surgery, the probability of relapse is high at 50–60%11 and there is a need for effective adjuvant therapy to improve survival.12 Based on the BILCAP study,13 international guidelines currently recommend adjuvant chemotherapy with capecitabine for 6 months as standard after resection.12 The ACTICCA-1 study comparing capecitabine with combination of cisplatin and gemcitabine (CISGEM) as adjuvant therapy following resection is ongoing.14 Patients with an early diagnosis of pCCA may be put forward for neoadjuvant chemotherapy and radiotherapy followed by liver transplant,7,15 but firm evidence of the survival benefit with such complex multimodal therapy is lacking.
Most patients with CCA have locally advanced (unresectable) or metastatic disease at presentation.9 Patients with unresectable, metastatic, or recurrent CCA receive palliative systemic therapies; those with locally advanced disease may also receive locoregional therapies.1 Chemotherapy is the mainstay of systemic treatment, with CISGEM being the current standard of care for first-line therapy.1,9,16 Alternative first-line regimens include CAPOX (capecitabine plus oxaliplatin, also known as XELOX)17 and GEMOX (gemcitabine plus oxaliplatin).18 Overall, palliative chemotherapy has very limited efficacy in BTC: median overall survival (OS) with CISGEM is <1 year.16 Published data suggest that 17.5–32.5% of patients who fail first-line therapy may be fit enough to receive second-line chemotherapy.19-22 Based on the findings of the ABC-06 trial,23 FOLFOX (folinic acid, fluorouracil, and oxaliplatin) can be considered the standard of care in this setting,9 although with modest efficacy (median OS: 6.2 months).23 There is currently no validated standard of care for third or further lines of therapy.
There is an urgent need for more effective treatment options for patients with unresectable, metastatic, or recurrent CCA. BTC have one of the highest frequencies of actionable molecular alterations among hard-to-treat solid tumours,24,25 so one potential route is via molecular testing to detect these alterations and open up the possibility of targeted, personalised medicine.
MOLECULAR TESTING USING NEXT-GENERATION SEQUENCING
Next-generation sequencing (NGS) encompasses several modern DNA and RNA sequencing technologies.26 It requires significantly less DNA or RNA than traditional Sanger sequencing, and is quicker, cheaper, and more accurate.27 A number of NGS platforms are commercially available (Table 1), and all are designed to screen for a wide panel of genetic alterations involved in cancer, including those described in CCA. Because of the ability of certain genes (e.g., FGFR2 and NTRK) to form fusions with multiple partners (described below), not all fusions are detected by DNA NGS platforms. Centres may therefore need to use additional RNA sequencing to obtain a more robust result, either through RNA NGS or other techniques. RNA NGS requires less sequencing than DNA NGS because of the intron splicing that occurs during mRNA production,28 and RNA sequencing has shown extremely high sensitivity and specificity in detecting actionable gene fusions.29 However, RNA is highly labile and can easily be damaged during preparation for the screen, leaving fragments that are too short to be informative.28 Some platforms offer both DNA and RNA NGS (Table 1); for example, the Oncomine™ Focus Assay (Thermo Fisher Scientific, Waltham, Massachusetts, USA) can screen both DNA and RNA in a single workflow.
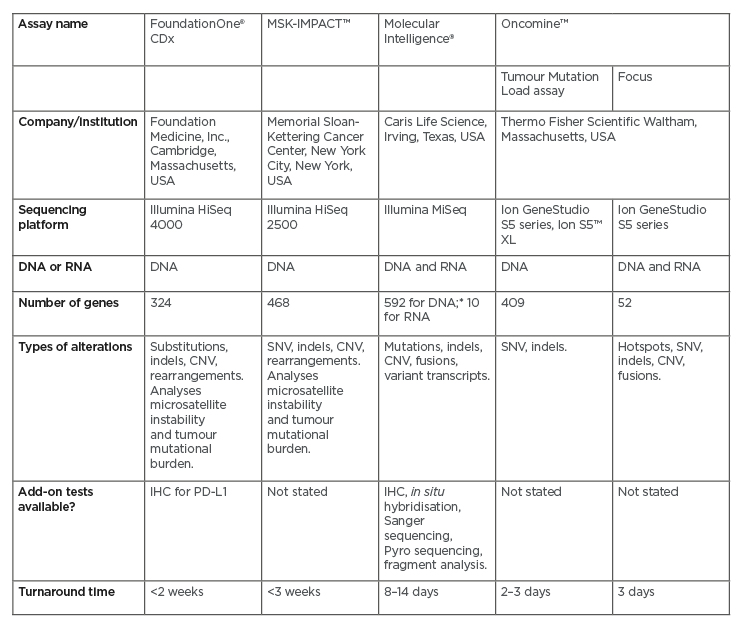
Table 1: Next-generation sequencing panels for genomic profiling of solid tumours.
The table shows a representative sample of the available next-generation sequencing platforms.
*Whole exome sequencing performed for all breast, ovarian, pancreas, and prostate cancers; all other tumours undergo 592-gene next-generation sequencing.
CNV: copy number variation; IHC: immunohistochemistry; SNV: single nucleotide variant.
Fluorescence in situ hybridisation and immunohistochemistry (IHC) are commonly used techniques that may be employed when NGS is unavailable. However, they are of limited use in characterising genetic aberrations, particularly fusions. Although both can detect the fusion gene, neither can further identify the fusion gene partner.30 Fluorescence in situ hybridisation can only detect a single target at a time; designing the multiple probes required to screen for every gene of interest is labour-intensive and not cost-effective.28,30
Guidelines on the use of molecular screening are becoming available; for example, in 2019 the European Society for Medical Oncology (ESMO) published recommendations on the use of NGS and other technologies for the detection of NTRK fusions.30 For cancers in which these fusions are rare, such as CCA, ESMO recommends using an NGS platform that is known to reliably detect these fusions and to include RNA testing where possible. IHC could then be used to confirm protein expression in positive cases.30 Where NGS is not readily available, ESMO recommends first using IHC to detect protein expression, and then to use NGS in cases where protein overexpression is detected. More recently, ESMO has recommended routine use of NGS in a number of advanced cancers, including CCA (described below).31
ACTIONABLE MOLECULAR TARGETS AND EMERGING TARGETED THERAPIES IN CHOLANGIOCARCINOMA
Molecular profiling using NGS has revealed the complex genetic makeup of the different CCA subtypes.32,33 For example, FGFR2 fusions and mutations in IDH1, IDH2, and BRAF genes are most frequent in iCCA, whereas HER2 (ERBB2) mutations and amplifications are predominantly found in pCCA and dCCA, as well as gallbladder cancers. KRAS and TP53 mutations are common in all subtypes.9,32,33 The distribution of NTRK mutations between CCA subtypes has not yet been reported.
The identification of these potentially actionable targets in CCA is of great clinical significance and has driven clinical trials investigating specific agents. Here, the authors highlight Phase II/III studies of the most promising agents directed at the key actionable molecular targets: FGFR2, IDH, NTRK, BRAF, and HER2. Immunotherapies are also briefly discussed.
Treatments Targeting FGFR2
The FGFR2 pathway is involved in cellular migration, proliferation, differentiation, and survival.34 50% of patients with CCA have a clinically significant genomic abnormality, such as an FGFR2 fusion or rearrangement.35 They are constitutively active and can occur with multiple partners.34-37 The most common FGFR2 fusions include FGFR2–PPHLN1, FGFR2–AHCYL1, and FGFR–BICC1.36 NGS has allowed the identification of novel FGFR2 fusions.38,39 For example, in an analysis of 118 FGFR2-positive patients enrolled in the Phase II FIGHT-202 study, Hollebecque et al.39 observed 54 unique FGFR2 rearrangements, of which 40 (74%) were unique to a single patient.
Pemigatinib is an FGFR1-3 selective kinase inhibitor that has recently received accelerated U.S. Food and Drug Administration (FDA) approval for treatment of adult patients with previously treated, unresectable locally advanced, or metastatic CCA with FGFR2 fusions or other rearrangements as detected by an FDA-approved NGS platform.40 This approval was based on the results of the FIGHT-202 study (Table 241-58), an international, multicentre, open-label, single-arm, multicohort, Phase II study in patients with CCA whose disease had progressed following at least one previous treatment and who had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0–2.41,42 In this study, patients were assigned to one of three cohorts depending on FGF/FGFR status: patients with FGFR2 fusions or rearrangements, patients with other FGF/FGFR alterations, or patients with no FGF/FGFR alterations. They received oral pemigatinib at a starting dose of 13.5 mg once daily (21-day cycle; 2 weeks on, 1 week off) until disease progression, unacceptable toxicity, withdrawal of consent, or physician decision. The primary endpoint was the proportion of patients with FGFR2 fusions or rearrangements who achieved an objective response, assessed by independent central review. A total of 146 patients were enrolled: 107 with FGFR2 fusions or rearrangements, 20 with other FGF/FGFR alterations, 18 with no FGF/FGFR alterations, and one with an undetermined FGF/FGFR alteration. The median follow-up was 17.8 months (interquartile range: 11.6–21.3). There were 38 (35.5%; 95% confidence interval [CI]: 26.5–45.4) patients with FGFR2 fusions or rearrangements who achieved an objective response (three had complete responses and 35 had partial responses). The median duration of response was 7.5 months, and median progression-free survival (PFS) was 6.9 months.42 FGFR–BICC1 was the most common fusion (present in 30% of patients); however, there were no meaningful differences in objective response rate (ORR), median PFS, or median OS between patients with FGFR–BICC1 fusions and those with other FGFR2 rearrangements.39 Hyperphosphataemia was the most common adverse event across all three cohorts, which was reported by 88 (60%) of 146 patients.42 Grade 3 or worse adverse events occurred in 93 (64%) patients, with the most frequent being hypophosphataemia (18 patients [12%]) and arthralgia (nine [6%]). Sixty-five (45%) patients had serious adverse events, with the most frequent being abdominal pain (seven [5%]) and pyrexia (seven [5%]). Overall, 71 (49%) patients died during the study; the most common cause of death was disease progression (61 [42%]) and no deaths were treatment related. The results of FIGHT-202 showed the therapeutic potential of pemigatinib in previously treated patients with CCA with FGFR2 fusions or rearrangements. Pemigatinib is currently being compared with CISGEM as first-line therapy for patients with unresectable and/or metastatic CCA in the Phase III FIGHT-302 study (Table 2).43
The FGFR inhibitors infigratinib and futibatinib (TAS-120) have also been shown to be effective in Phase II studies in pretreated patients with FGFR2 alterations, with manageable toxicity (Table 2).44,45,47,48 Corresponding Phase III studies in patients with previously untreated advanced CCA are ongoing.46,49 Other FGFR inhibitors are also under investigation in Phase II studies (Table 2).50-53
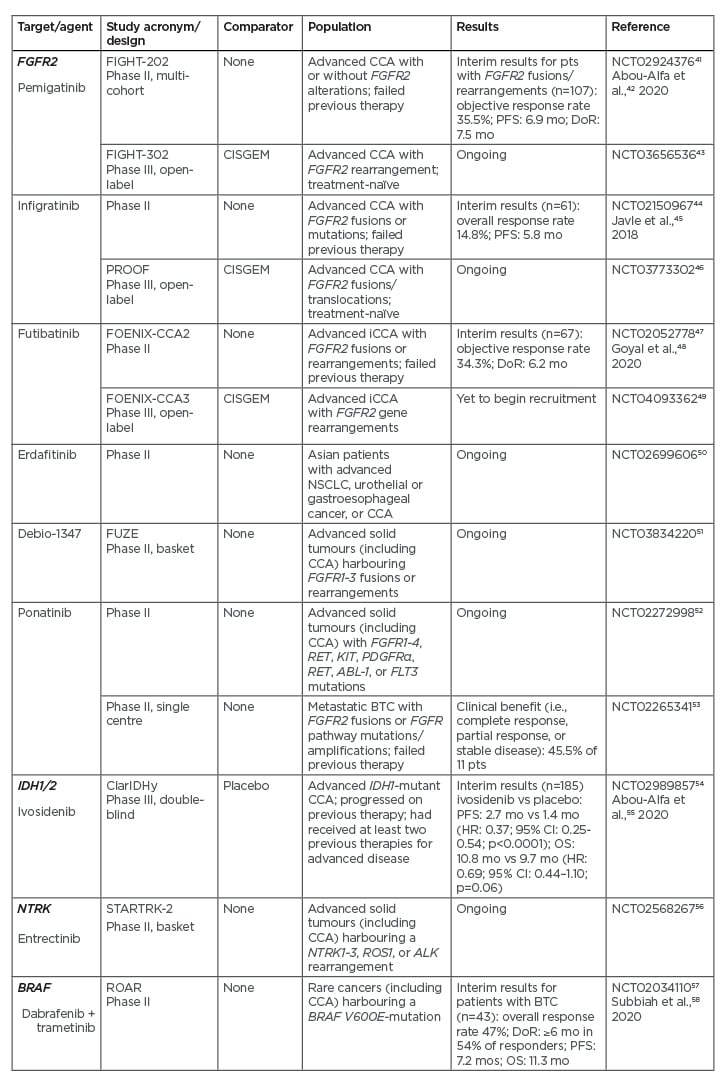
Table 2: Recent or ongoing Phase II/III studies of treatments targeting the key driver mutations FGFR2, IDH, NTRK, and BRAF in cholangiocarcinoma.
CCA: cholangiocarcinoma; CI: confidence interval; DoR: median duration of response; HR: hazard ratio; mo: months; NSCLC: non-small cell lung cancer; OS: median overall survival; PFS: median progression-free survival; pts: patients; vs: versus.
Treatments Targeting IDH
Mutations in IDH1 and IDH2 are associated with the production of the aberrant, oncogenic metabolite, 2-hydroxyglutarate.59 IDH1 mutations are present in approximately 13% of patients with iCCA, compared with 0.8% of patients with pCCA/dCCA.60 ClarIDHy is an international, randomised, placebo-controlled Phase III study of the IDH1 inhibitor ivosidenib in 185 patients with advanced, pretreated CCA with IDH1 mutations who had progressed on previous therapy, and had up to two previous treatment regimens for advanced disease and an ECOG PS score of 0 or 1 (Table 2).54,55 The primary endpoint was PFS, assessed by an independent central review.
An interim analysis showed that treatment with ivosidenib 500 mg once daily resulted in a significant improvement in median PFS over placebo (2.7 months versus 1.4 months; hazard ratio [HR]: 0.37; 95% CI: 0.25–0.54; p<0.0001).55 Median OS was 10.8 months with ivosidenib versus 9.7 months with placebo (HR: 0.69; 95% CI: 0.44–1.10; p=0.060). The study, however, allowed patients in the placebo group to cross over to the ivosidenib group at disease progression. Adjustment for this gave a median OS of 6.0 months for the placebo group, which was significantly shorter than for the ivosidenib group (HR: 0.46; 95% CI: 0.28–0.75; p=0.0008).55 More mature data regarding OS are awaited. The most common Grade 3 or worse adverse event in both treatment groups was ascites, which occurred in four (7%) of 59 patients receiving placebo and nine (7%) of 121 patients receiving ivosidenib. Thirty-six (30%) patients receiving ivosidenib and 13 (22%) patients receiving placebo had serious adverse events. There were no treatment-related deaths. The results of ClarIDHy support the clinical benefit of targeting IDH1 mutations in advanced, IDH1-mutant CCA.
Treatments Targeting NTRK
The NTRK1, NTRK2, and NTRK3 genes encode the receptor tyrosine kinases TRKA, TRKB, and TRKC, which are pivotal in the development and function of the nervous system.61 NTRK fusions are rare in CCA: recent studies suggest they are present in <1% of patients.62,63 They are better characterised in Asian patients than in Caucasian patients64 and the development of specific NTRK inhibitors means that interest in this mutation is growing. Like FGFR2, NTRK can partner with a variety of other genes,38,65 and novel fusions have been identified through NGS.38
The NTRK inhibitor larotrectinib is approved in Europe and the USA for patients with solid malignancies and a proven NTRK gene fusion without a known acquired resistance mutation.66,67,68 In a Phase I/II study in 55 adults and children who had tumours with these fusions, the overall response rate (primary study endpoint) was 75% (95% CI: 61–85%) according to an independent review, including one of the two patients in the study with CCA.68 At 1 year, 71% of the responses were ongoing and 55% of the patients remained progression-free. Toxicity was mild, with most adverse events being Grade 1.
Entrectinib has received FDA breakthrough designation status for treatment of cancers harbouring NTRK69 and is currently being evaluated in the Phase II STARTRK-2 basket study (Table 2).56
Although NTRK fusions are rare in CCA, and CCA are uncommon in most Western countries, the marked and durable antitumour activity of NTRK inhibitors in patients with NTRK fusion–positive cancer, regardless of the age of the patient or the tumour type, warrants adding these targeted agents to the therapeutic armamentarium of CCA.
Treatments Targeting BRAF
Mutations in BRAF lead to constitutive activation of the mitogen-activated tyrosine kinase pathway, resulting in increased cell proliferation and decreased apoptosis.70 The most common BRAF mutation is V600E, which occurs in approximately 5% of patients with iCCA.71,72 The combination of dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) has shown activity in several BRAF V600E-mutated cancers. In a study that forms part of an ongoing, Phase II, open-label, single-arm, multicentre, Rare Oncology Agnostic Research (ROAR) basket trial in patients with BRAF V600E-mutated rare cancers, 43 patients with BRAF V600E-mutated, advanced BTC (iCCA, 39 patients), an ECOG PS of 0–2, and who had received previous systemic treatment were treated with oral dabrafenib 150 mg twice daily and oral trametinib 2 mg once daily until disease progression or intolerance of treatment.58 The overall response rate (the primary study endpoint) was 47% (95% CI: 31–62%). The most common Grade 3 or worse adverse event was increased γ-glutamyltransferase level, which was reported by five (12%) patients. Seventeen (40%) patients had serious adverse events; these were treatment-related in nine (21%) patients. The most frequent treatment-related serious adverse event was pyrexia, which occurred in eight (19%) patients. These results support consideration of dabrafenib plus trametinib combination treatment in patients with BRAF V600E-mutated BTC. The study authors recommend that routine testing for BRAF V600E mutations should be considered in all patients with BTC.
Treatments Targeting HER2
Overexpression of HER2 is a key driver of tumour development in several cancers.73 A number of targeted treatments are already available for HER2-positive patients with breast cancer, non-small cell lung cancer, gastric cancer, and pancreatic cancer. These include the tyrosine kinase inhibitors lapatinib, erlotinib, and afatinib, and the anti-HER2 antibodies trastuzumab and pertuzumab. Given this, HER2 is considered a candidate for targeted therapy in CCA. Presently, evidence is lacking, but preclinical studies suggest that the antibody-cytotoxic drug conjugate trastuzumab emtansine and the dual EGFR/HER2 inhibitor NVP-AEE788 may be effective in BTC.74,75 There are also isolated reports of HER2-positive CCA patients responding to treatment with targeted therapy.76,77
HER2 targeting may benefit patients with HER2 amplifications rather than those with mutations.77 HER2 amplifications are rarely seen in iCCA, so HER2-directed treatment may benefit patients with pCCA/dCCA more than those with iCCA.78
Immunotherapies in Cholangiocarcinoma
Upregulation of immune checkpoint molecules has been shown in CCA,32 making immunotherapy another area of interest for targeted treatment. The antiprogrammed death-1 antibody pembrolizumab is being investigated as single-agent therapy in pretreated patients with advanced BTC in the ongoing Phase II KEYNOTE-15879 and Phase Ib KEYNOTE-02880 studies. Analysis of data from KEYNOTE-158 (n=104) revealed an overall response rate of 5.8% and a median OS of 7.4 months.81 Pembrolizumab is also being investigated as combination therapy with CISGEM.82
Another antiprogrammed death-1 antibody, nivolumab, is also under investigation as a treatment for CCA. A small Phase I study in Japan showed encouraging efficacy for nivolumab both as monotherapy (median PFS: 1.4 months; median OS: 5.2 months) and in combination with CISGEM (median PFS: 4.2 months; median OS: 15.4 months).83 In a Phase II study, 45 patients with advanced BTC who received nivolumab as monotherapy had a median PFS of 3.98 months and a median OS of 14.22 months.84,85 Nivolumab is currently being investigated in a Phase II study as combination therapy with either CISGEM or the anticytotoxic T-lymphocyte-associated protein 4 antibody ipilimumab in advanced BTC,86 and in combination with ipilimumab in two basket studies.87,88 A subgroup analysis of data from 39 patients with BTC enrolled in one of these basket studies88 has been published: the ORR was 23%, with a median PFS of 2.9 months and a median OS of 5.7 months.89
The combination of the antiprogrammed death ligand-1 antibody durvalumab and the anticytotoxic T-lymphocyte-associated protein 4 antibody tremelimumab has shown promising efficacy when given alongside CISGEM in treatment-naïve patients with advanced BTC:90 an interim analysis revealed an ORR of 73.3%, a median PFS of 11.9 months, and a median OS of 20.7 months.91 Durvalumab is also currently being evaluated in combination with CISGEM in the Phase III TOPAZ-1 trial.92
PROPOSED CLINICAL WORKUP FOR PATIENTS WITH UNRESECTABLE CHOLANGIOCARCINOMA
Figure 1 shows how molecular testing should fit into the clinical pathway once CCA has been diagnosed via imaging and histology.
The authors propose that all patients with unresectable CCA should undergo molecular testing at the time of initial diagnosis. A combination of DNA NGS and RNA sequencing (either via NGS or other techniques) should be used. In addition, patients who have already been diagnosed and are receiving CISGEM could be tested at the point of disease progression to determine whether they have a mutation that would allow enrolment into a clinical trial for those who have failed previous therapy. If a patient fails targeted therapy, NGS could be used to determine whether a resistance mutation is present.
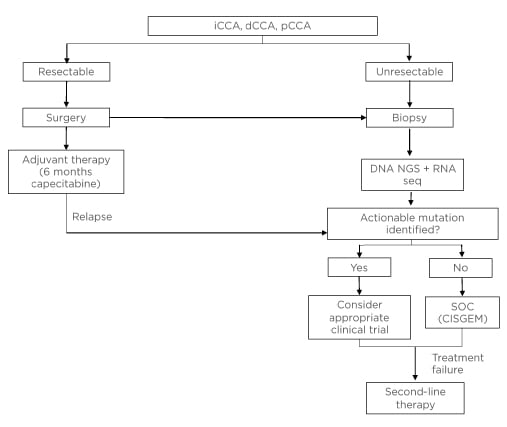
Figure 1: Proposed clinical workup for patients with unresectable or metastatic cholangiocarcinoma.
CISGEM: cisplatin plus gemcitabine; dCCA: distal cholangiocarcinoma; iCCA: intrahepatic cholangiocarcinoma; NGS: next-generation sequencing; pCCA, perihilar cholangiocarcinoma; seq: sequencing; SOC: standard of care.
Given the high incidence of tumour recurrence among patients who undergo surgery, molecular testing might also be carried out for these patients so that the appropriate course of treatment can be implemented in the event of relapse. However, whether molecular profiling at the time of surgery reliably reflects the molecular contexture at the point of recurrence is currently unclear.
Unfortunately, not all patients are currently able to benefit from molecular testing. In some European countries, such as France, molecular profiling is only available through institutional programmes or clinical trials. In addition, reimbursement varies across Europe. For example, in France there is no reimbursement for molecular testing, and it is not available privately. In Switzerland, NGS is reimbursed, but reimbursement for molecular targeted medications may not yet be established. This means that if a clinician wants to prescribe a particular targeted therapy, individual approval by the patient’s health insurer needs to be obtained. It is important that barriers to molecular testing and subsequent targeted treatment are removed, so that all eligible patients can be considered for personalised therapy.
The data generated by NGS and other molecular profiling techniques can be complex. A molecular tumour board, with experts such as molecular oncologists, clinical geneticists, and molecular pathologists, can help clinicians interpret the data and suggest treatment options.93,94 A multidisciplinary oncology board that includes an oncologist, hepatologist, pathologist, radiologist, radiation therapist, surgeon, nurse specialist, and primary care physician should then be involved in discussion of diagnosis and treatment. Research has shown that these boards have several benefits, including improved diagnostic decision-making, enhanced care, and knowledge transfer between teams.94-96
ESMO recently recommended routine use of NGS on tumour samples in CCA, advanced non-squamous, non-small cell lung cancer, prostate cancers, and ovarian cancers.31 Other recommendations include using off-label drugs matched to genomics only when a national or regional access programme and decision procedure is in place, and that research centres develop multigene sequencing as a tool to screen patients eligible for clinical trials and to accelerate drug development. Data that could inform optimisation of the technology should be prospectively captured.
CONCLUSION
There has traditionally been a severe lack of treatment options for patients with unresectable CCA. However, there are now several promising emerging therapies that target driver mutations and may pave the way for a more personalised treatment approach. It is therefore important that patients undergo molecular screening at the time of initial diagnosis so that instead of standard of care chemotherapy, they are offered the option to enrol in an appropriate clinical trial, thereby giving them access to a more promising treatment while expanding the knowledge of the genetic makeup of CCA. Patients educated in the process of their own care and multidisciplinary oncology teams should collaborate as ‘partners’, so that the proposed algorithm for unresectable CCA can be practically applied for many patients in need. This may be the first step in overcoming barriers to performing molecular testing and using novel targeted therapies, prior to starting standard chemotherapy, whenever possible, in this aggressive malignancy.






