
Abstract
Myelodysplastic syndrome (MDS) is a heterogeneous group of myeloid disorders. Allogeneic stem cell transplantation (alloSCT) is the therapeutic approach with a known curative potential for patients with MDS, which allows long-term disease control to be achieved. Despite advances in transplantation technology, there is still a considerable morbidity and mortality associated with this approach. Moreover, numerous controversies still exist regarding alloSCT in MDS. There is significant variability in the management of patients with MDS, especially of the intermediate-risk category and specifically in regards to the timing and use of transplantation. Modern genetic analysis has identified a variety of new mutations, which are associated with clinical phenotype and prognosis. Whether somatic mutations are important prognostic markers of response to alloSCT is little known. It is not clear whether somatic mutations can help to identify groups that are most likely to benefit from alloSCT. In this article, we review the current status of somatic mutations in MDS and focus on the prognostic impact of mutations in the context of alloSCT.
INTRODUCTION
Myelodysplastic syndromes (MDSs) are a clinically and molecularly heterogeneous group of clonal stem cell disorders characterised by ineffective and dysplastic myeloid cell differentiation and a high rate of progression to acute myeloid leukaemia (AML). Treatment options for MDS range from observation and growth-factor therapy to more intensive approaches, such as allogeneic stem cell transplantation (alloSCT).1,2 AlloSCT is the only therapeutic option that has the potential to produce long-term remission and disease-free survival.3 Furthermore, introduction of reduced intensity conditioning decreased non-relapse mortality, especially in older patients.4 Nevertheless, alloSCT is a high-risk procedure that is often associated with severe complications.
The main cause of treatment failure in MDS patients after alloSCT is relapse of disease. Although significantly reduced over the last 20 years, mortality and morbidity risks associated with alloSCT continue to represent a major limit to feasibility in a large number of patients.5 The international prognostic scoring system and the revised version (IPSS-R) are usually used to determine risk stratification.6,7 Recently it was demonstrated that alloSCT offers optimal survival benefit when it is performed early in the intermediate-risk stage.8 Other studies have shown that poor and very poor-risk of cytogenetics within the IPSS-R cytogenetic risk categories are strong predictors of adverse outcomes after alloSCT, especially when other risk factors are considered simultaneously.7
Further disease characteristics such as peripheral cytopenias and a high percentage of blasts in the bone marrow have been shown to affect the prognosis of patients with MDS after alloSCT. Moreover, age-related factors significantly affect the risk of the non-relapse mortality.9,10 Further successful treatment with hypomethylating agents has been shown to be associated with better outcome after alloSCT.11 Due to the fact that MDS has a highly variable course of disease, accurate risk stratification and prognosis becomes crucial for therapeutic decision-making. Selection of patients and choosing the optimal timing of transplantation are areas which require further research to improve use of alloSCT in MDS.
The mutation status of a growing number of genes has recently been found to be an important prognostic factor in MDS patients.2,4,12-14 Although the prognostic value of mutations has been well studied, little is known about the predictive impact of specific genetic lesions in MDS patients on outcome after alloSCT. In this review, we focus on the prognostic impact of somatic mutations in MDS patients in the context of alloSCT.
MUTATION IN MYELODYSPLASTIC SYNDROMES
Phenotypic heterogeneity of MDS reflects different somatic mutations that cause clonal proliferation and evolution. High-throughput molecular technologies, such as next generation sequencing and high-density single nucleotide polymorphism arrays, provide opportunities for patient stratification and personalised treatment based on individual mutation profiles. Recent genetic analyses have identified a variety of new mutations which present at distinct frequencies in subgroups of patients with MDS.2,14-17 With the advent of next-generation sequencing recurrent somatic mutations are observed in >90% of patients with MDS, with the number of driver mutations having an independent prognostic impact.2,4,18,19 Mutations in several genes have been reported to influence overall survival (OS) and risk of disease progression.2,4,12-14 Knowledge about the nature of genes involved in the mutations may improve the understanding of the evolution and development of MDS.14 In MDS, somatic mutations affect specific classes of genes. The most common mutations detected in MDS patients include RNA splicing machinery, DNA methylation, chromatin modification, transcription regulation, and DNA damage response (Table 1).
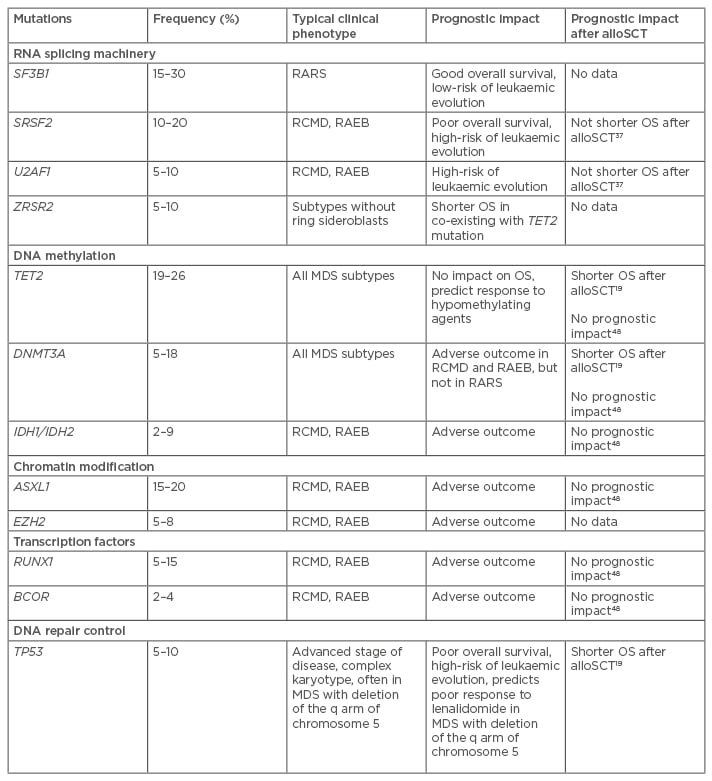
Table 1: Most common somatic mutations in myelodysplastic syndrome patients.
OS: overall survival; MDS: myelodysplastic syndrome; alloSCT: allogeneic stem cell transplantation; RARS: refractory anaemia with ring sideroblasts; RCMD: refractory cytopenias with multilineage dysplasia; RAEB: refractory anaemia with excess blasts.
RNA SPLICING MACHINERY
The editing process during transcription of DNA into RNA, which involves removal of non-coding regions, introns, is called splicing.20 RNA splicing is the most frequently mutated pathway in MDS. The most commonly affected spliceosome genes included SF3B1 (splicing factor 3b subunit 1), U2AF1 (U2 small nuclear RNA auxiliary factor 1), U2AF2 (U2 small nuclear RNA auxiliary factor 2), SRSF2 (serine/arginine-rich splicing factor 2), ZRSR2 (zinc finger [CCCH type], RNA binding motif, and serine/arginine rich 2), and some others.16,18,21-24 Splicing factor mutations, particularly in SF3B1, U2AF1, SRSF2, and ZRSR2, are present in 50–65% of MDS cases, making them the most common class of mutations in MDS.18,21,24-26 Mutations in the other spliceosome genes (U2AF2, SF1, PRPF40B, and SF3A1) occurred with a frequency of ≤1%.21,23,25,27,28
A growing body of evidence demonstrates that splice gene mutations define distinct clinical phenotypes and show preferential associations with mutations targeting transcriptional regulation. Recent findings support the role of spliceosome mutations as early mutations that interact with other clonal and sub-clonal events within the same genes during leukaemogenesis in patients with MDS.29
SF3B1
SF3B1, together with other factors, forms the U2 small nuclear ribonucleoproteins complex and binds pre-mRNA. Recurrent mutation of the SF3B1 gene was found in 20–35% of MDS patients and in 70% of MDS patients whose disease is characterised by the presence of ring sideroblasts (in refractory anaemia with ring sideroblasts [RARS]),4,18,21,22,30,31 although the clonal dominance of mutations in granulocytic cells suggests that oncogenic effects may not be restricted to the erythroid lineage.21 It was noted that SF3B1 mutations were frequently associated with an isolated deletion of the q arm of chromosome 5.18 SF3B1 mutations are associated with a better clinical outcome.4 There are no data about the predictive impact of this mutation on outcome after alloSCT. Given the frequent combination of SF3B1 mutation with favourable prognostic markers and with good clinical outcome, the presence of SF3B1 mutations as a single aberration should not be an indication for alloSCT.
SRSF2
The protein SRSF2 forms part of the spliceosome. This protein is important for splice-site selection, spliceosome assembly, and both constitutive and alternative splicing.32 SRSF2 is currently the second most frequently mutated splicing gene in the myeloid diseases.18,33 These genetic aberrations are almost always heterozygous missense mutations that specifically occur at the amino acid residue P95. In MDS, an SRSF2 mutation was detected in approximately 10% of cases. They are most often observed in refractory cytopenia with multilineage dysplasia (RCMD) and refractory anaemia with excess blasts (RAEB), two subtypes of high-risk MDS.13,27 In chronic myelomonocytic leukaemia (CMML), SRSF2 is the most commonly mutated spliceosome gene (28–47%) and has been associated with older age, less pronounced anaemia, and a normal karyotype.25,33
MDS harbouring an SRSF2 mutation is associated with a higher rate of transformation to AML and with lower OS compared with MDS with wild-type SRSF2.30 However, more recently, the frequency of complex karyotypes and adverse cytogenetic risk aberrations were found to be higher in patients with SRSF2 mutations than in MDS patients with other mutations.29 Although this mutation was found to be associated with a poor prognosis,30 significant difference in OS between wild-type and mutated SRSF2 in transplanted patients was not detected.34
U2AF1
U2AF1 encodes the auxiliary factor for the U2 pre-mRNA splicing complex. Eleven distinct mutations have been reported in the U2AF1 gene.28,30,35,36 Most of these mutations occur within the two zinc finger domains of U2AF1, with S34 and Q157 residues.34 Using mouse models it was shown that U2AF1 mutations drive further mutation.37 In MDS, U2AF1 mutations are seen in <10% of patients. Although U2AF1 mutations have been associated with less favourable OS and a higher risk of transformation to AML,13,23 it is not clear whether the mutation has independent prognostic impact.25,29 The prognostic effect is known to depend on certain biological factors as well as a combination of cytogenetics and other mutations.
Both the U2AF1 and SRSF2 mutations have been associated with poor outcome.23 Recently, two groups reported no difference in survival between mutated splicing genes and wild-type Wilms’ tumour patients undergoing alloSCT.19,34 Despite some limitations, such as a heterogeneous cohort of patients, these studies provide preliminary evidence to support the theory that alloSCT may potentially reduce the effects of certain poor-risk somatic mutations recurring in MDS.34
ZRSR2
ZRSR2 encodes an essential splicing factor which functions in early spliceosome assembly through direct interactions with U2AF2.38 Mutations of the ZRSR2 gene are present in 10–15% of MDS cases.18,28 In contrast to other spliceosome gene mutations, ZRSR2 mutations occur across the entire length of the transcript. Mutations in ZRSR2 are more prevalent in MDS subtypes without ring sideroblasts and CMML.25,28 Some authors have shown that ZRSR2 mutations are associated with an elevated percentage of bone marrow blasts and higher rates of progression to AML.25,28 However, this was not confirmed by other studies. Furthermore, it was shown that the prognostic impact of ZRSR2 mutations depends on the mutation status of TET2, the gene most commonly combined with ZRSR2 mutations.30 There are no data about the prognostic impact of ZRSR2 mutations on outcome after alloSCT.
DNA METHYLATION
Epigenetic regulation refers to methylation of cytosine residues of DNA, biochemical modifications of histones, and the expression of non-coding RNAs.39 Mutations in the epigenetic modifiers are of particular importance in MDS.
Ten-Eleven Translocation
The ten–eleven translocation (TET) protein family is responsible for demethylation.40 In addition, TETs regulate gene expression through modification of chromatin at promoter regions.41 TET2 is a tumour suppressor gene, and so loss-of-function mutations support the abnormal haematopoiesis observed in MDS.42 In MDS, TET2 mutations affect 19–26% of MDS patients.43 TET2 mutations are observed in all types of MDS.13,44 The clinical implication of TET2 mutations remains unclear. TET2 mutations are a neutral or favourable prognostic biomarker44,45 and have been shown to predict response to hypomethylating therapy in MDS.43 Some studies demonstrated a higher overall response rate to azacitidine treatment in TET2 mutated cases with no difference in OS.46,47
In MDS patients following alloSCT, TET2 mutations are associated with shorter OS. Taking into account other risk factors, such as complex karyotype, high level of bone marrow blasts, donor type, and conditioning regimen, TET2 mutations together with TP53 and DNMT3A, are significantly associated with shorter OS.19 However in another study, TET2 mutations showed no prognostic impact in MDS patients uniformly treated with FLAMSA-busulfan conditioning.48
DNMT3A
DNMT3A (DNA methyltransferases 3A) together with other methyltransferases conducts de novo methylation of cytosine residues in CpG islands.49,50 DNMT3A mutations occur in approximately 5–18% of MDS patients with the most common mutation involving R882 amino acid residue.19,43,51,52 These mutations can be found in all MDS subtypes.13 DNMT3A mutations are associated with older age at diagnosis but not with other clinical features or cytogenetics.52 In RARS, DNMT3A mutations more commonly co-operate with SF3B1 mutations.13
Some studies have shown an adverse clinical outcome with a low survival rate and rapid progression to AML in MDS patients with DNMT3A mutations.52 Moreover, it was found that DNMT3A mutations have been associated with poor outcomes in some types of MDS, particularly in RCMD and RAEB.13,52 In RARS, DNMT3A mutations often co-occur with SF3B1 mutations and have no association with adverse outcome.13,53
Bejar et al.19 have identified this mutation as a risk factor, which significantly reduces the OS after alloSCT. The authors observed an increased risk of relapse and death after transplantation, particularly when other predictive variables were considered.19 In contrast, Christopeit et al.48 showed no prognostic impact of DNMT3A mutations in patients after alloSCT.
IDH1 and IDH2
IDH1 and IDH2 genes encode cytoplasmic/peroxisomal isocitrate dehydrogenase 1 and mitochondrial isocitrate dehydrogenase 2, respectively. These are NADP+-dependent enzymes that catalyse the oxidative decarboxylation of isocitrate to -ketoglutarate, generating NADPH from NADP+. Both genes function in a crossroads of cellular metabolism, cellular defence against oxidative stress, oxidative respiration, and oxygen-sensing signal transduction.54 In MDS, somatic mutations in IDH1/2 occur in 2–9% of patients.45,55-58 Mutations preferentially affected the IDH2 gene. Since these mutations were discovered in early MDS, prior to AML transformation, it has been suggested that IDH mutations, especially IDH2, may contribute to the pathogenesis of MDS.56
The simultaneous presence of IDH mutations with other genetic changes is commonly observed in myeloid neoplasms.57 The close association of IDH2 mutations with ASXL1, DNMT3A, and SRSF2 suggests that IDH2 mutations can interact with these aberrations in the development of MDS.57 The prognostic implication of IDH mutations in MDS remains unclear. Some studies have shown an adverse clinical outcome in MDS patients with IDH mutations.45,58 Furthermore, two studies demonstrated IDH1 mutation as an independent predictor of inferior survival.55,59 Very little is known about the prognostic impact of IDH1/2 mutations in MDS patients after alloSCT. Only one study has examined this relationship and suggests that there is no prognostic significance of these mutations.48
CHROMATIN MODIFICATION
Two genes involved in chromatin modification and regulation are recurrently mutated in MDS: ASXL1 (additional sex combs like 1) and EZH2 (enhancer of zeste 2 polycomb repressive complex 2), which encode chromatin-modifying proteins and interact with the polycomb-group repressive complex 1 and 2. Genomic loss of EZH2 contributes to overexpression of the Hox gene clusters in MDS through epigenetic modifications.60 Both mutations are most often observed in higher-risk subtypes of MDS such as RCMD and RAEB.13,61
ASXL1 and EZH2
ASXL1 and EZH2 mutations are closely associated with myeloproliferative disorders. In MDS, ASXL1 mutations are found in 15–20% of patients.45,61 These mutations are generally associated with signs of aggressive progression and poor clinical outcome.45 Mutations of EZH2 occur in 5–8% of MDS patients.13,45 Previously, it was reported that EZH2 mutations are a prognostic biomarker, associated with shorter OS.45 Although there are no reports on the effect of ASXL1 and EZH2 mutations on outcome after alloSCT, the overall relationship of these mutations with poor outcome may be important for risk stratifications.
TRANSCRIPTION REGULATION
Runt-related transcription factor 1 (RUNX1) protein takes part in regulating the expression of multiple genes involved in normal haematopoiesis.62,63 RUNX1 mutations occur in 5–15% of MDS patients,18,64 and are most often observed in RCMD and RAEB.13 RUNX1 mutations are detected during disease progression. Furthermore, this mutation is significantly increased even in secondary AML following MDS. The acquisition of additional mutations in genes such as MLL-PTD or FLT3-ITD with RUNX1 appears to be an event precipitating leukaemic transformation.65 RUNX1 mutations are associated with unfavourable clinical outcomes.45 Little is known about the prognostic impact of this mutation after alloSCT. In one cohort of MDS patients uniformly treated with FLAMSA-busulfan conditioning, RUNX1 mutations showed no prognostic impact.48
BCOR (BCL6 corepressor) and BCORL1 (BCL6 corepressor-like 1) code for related transcriptional corepressors, that are found tethered to promoter regions by DNA-binding proteins. The proteins interact with several different Class II histone deacetylases to repress transcription. Recently, sequencing of BCOR genes in MDS patients identified 2–4% of mutations.66 BCOR mutations are often associated with RUNX1 and DNMT3A mutations. These data suggest that BCOR mutations define the clinical course rather than disease initiation.66,67 In MDS, BCOR mutations are associated with adverse outcome, shorter OS, and higher incidence of AML transformation.66
DNA REPAIR CONTROL
Tumour protein p53 (TP53) is a tumour suppressor protein that regulates expression of genes involved in cell cycle arrest, apoptosis, senescence, DNA repair, and changes in metabolism.68 TP53 is the most frequently mutated gene in many types of cancer. In MDS, TP53 mutations occur in 5–10% of cases.13,45 These mutations are most often observed in patients with advanced disease, complex karyotype, or adverse karyotype aberrations.13,45 MDS patients carrying TP53 mutation have an unfavourable clinical outcome and a high risk of leukaemic evolution.4,45
Bejar et al.19 have analysed somatic mutations as a potential risk factor in MDS patients following alloSCT. In this analysis, only mutations in TP53 were significantly associated with shorter OS and progression-free survival. TP53 mutations are closely associated with complex karyotype. However, in patients with complex cytogenetics, TP53 mutation showed significantly shorter OS. In contrast, patients with complex cytogenetics without TP53 mutation had OS comparable to that of patients without complex cytogenetics.19 Moreover, there are data demonstrating that TP53 mutations in low-risk MDS with deletion of the long arm of chromosome 5 treated with lenalidomide are associated with an increased risk of leukaemic evolution.69 In view of the above, this group of low-risk MDS patients could be candidates for alloSCT.
CONCLUSIONS
Currently, little is known about the prognostic value of mutations in MDS patients in the context of alloSCT. Several groups studied the role of alloSCT to overcome the negative impact of mutations in MDS patients. Bejar et al.19 have found that TP53, TET2, and DNMT3A mutations are significantly associated with shorter OS following alloSCT. Importantly, the authors observed no adverse prognostic signs such as cytogenetics or bone marrow blasts percentage before transplantation in DNMT3A and TET2 mutated cases. Nevertheless, after alloSCT these patients had an increased risk of relapse and death, particularly when other predictive variables were considered. In this study, TP53 mutation status was the most significant predictor of mortality after transplantation.
Christopeit et al.48 analysed MDS patients treated with FLAMSA-busulfan sequential conditioning and alloSCT. RUNX1, GATA2, TET2, and CEBPA and TP53 mutations were the most frequently observed in this cohort. None of the mutations showed a prognostic impact in this analysis. Recently, Hamilton et al.37 examined the effect of alloSCT on the negative prognostic impact of U2AF1 and SRSF2 mutations in MDS patients. They reported no difference in survival between mutated splicing genes and wild-type patients undergoing transplantation. Thus, the authors suggest that patients with such mutations could benefit from alloSCT.
All reports have some limitations, such as heterogeneity of patients, conditioning, and others. Despite these limitations, some analysis has provided preliminary evidence to support the hypothesis that alloSCT may mitigate the effects of certain poor risk somatic mutations. In general, somatic mutations such as TP53, TET2, and DNMT3A in MDS patients might be important predictors for adverse outcome after alloSCT.19,37
MDS is characterised by mutations in >40 genes. The complex structure of gene crosstalk, multiple mutations, and extensive sub-clonal architecture are common for MDS.4,25 Recent genetic analysis of large populations revealed some somatic mutations in healthy people in advanced age. These findings support the hypothesis that mutations in genes such as DNMT3A, ASXL1, TET2, and others are likely to be initiating events for MDS and/or AML.17,70-72 These mutations can confer an increased risk of subsequent haematological malignancy diagnosis as well as higher all-cause mortality.17 The discovery of these mutations in elderly patients with low-risk MDS should be considered in risk stratification very carefully.
Accurate risk stratification, patient selection, and timing of alloSCT are frequently difficult in MDS. The decision on whether or not alloSCT is indicated can be challenging. Integration of somatic mutations into prognostic scoring systems may facilitate risk stratification of individual patients and further refine clinical decision-making in MDS.






