Abstract
Inhibitors of poly(ADP-ribose) polymerase (PARP), such as olaparib and talazoparib, have recently been approved as therapies for BRCA-mutated human epidermal growth factor receptor 2 (HER2)-negative metastatic breast cancer (BC). In addition, olaparib, as well as rucaparib and niraparib, have received approval for treatment of patients with BRCA-mutated or platinum-sensitive recurrent ovarian cancer.
The treatment efficacy of PARP inhibitors is higher in case of malignancies that harbour deleterious germline or somatic BRCA mutations compared to BRCA wild-type tumours. Consequently, BRCA mutations or intrinsic tumour sensitivity to platinum therapy are considered indicators of impaired ability to repair DNA double-strand breaks via homologous recombination.
However, not all BRCA-mutated cancer patients benefit from PARP inhibitors. In contrast, for some patients with wild-type BRCA or platinum-resistant tumours, the PARP inhibitors may still offer some therapeutic advantages. Therefore, there is a need to determine additional biomarkers to more precisely select patients without deleterious BRCA mutations, who may be eligible for treatment with PARP inhibitors.
The main objective of this mini-review is to present the main mechanisms of action of PARP inhibitors and briefly summarise the clinical trials leading to their approval in treatment of BRCA-mutated, HER2-negative metastatic BC. In addition, this article discusses the efficacy, safety, and resistance to PARP inhibitors in women with metastatic BC.
Introduction
Physiologically, cells are equipped with DNA damage repair systems that can correct various errors. However, congenital defects in DNA repair mechanisms may cause accumulation of DNA mutations and elevated risk of malignancy. Certain genes that are involved in DNA damage repair pathways play a key role in neoplastic development. For instance, an elevated susceptibility to inherited breast cancer (BC) and ovarian cancer (OC) has been reported in women harbouring germline mutations of BRCA1 and BRCA2 (both tumour suppressor genes).1,2 Despite their structural differences, these genes perform similar cellular actions and are instrumental for genome protection.1,2 The BRCA1 gene is located on the long arm of chromosome 17, and BRCA2 gene on the long arm of chromosome 13.3 Currently, >2,000 mutations have been detected in both BRCA genes (e.g., deletions, insertions, or duplications) that result in various aberrant transcriptional outcomes (e.g., missense, nonsense, silent, and splice-site).3 From a clinical point of view, BRCA changes that augment cancer susceptibility have been recognised as deleterious mutations, and usually result in nonfunctional proteins.3 As a consequence, such mutations interfere with the repair of the damaged DNA. For instance, a large study on BRCA1 or BRCA2 mutation carriers has revealed a significant cumulative risk of BC and OC, in regard to both the gBRCA1m mutation (72% for BC and 44% for OC) and gBRCA2m mutation (69% for BC and 17% for OC).4 In contrast, in the general population of women the cumulative risk is only 12.0% for BC, and 1.3% for OC.5 Furthermore, malignancy risk differs depending on various predictors (e.g., family history) and the location of mutations within the BRCA1 and BRCA2 genes.4 It should be emphasised that deleterious BRCA1 or BRCA2 mutations can also augment the risk of breast and prostate cancer in men,6 as well as pancreatic and stomach cancers,7,8 or colorectal cancer, in both women and men.9
Poly(ADP-ribose) polymerases (PARP) are a family of enzymes that transfer adenosine diphosphate (ADP) ribose parts to surrounding proteins in response to different cellular stimuli. PARP1 and PARP2 participate in the cellular response to single-strand DNA breaks.10 Inhibition of the ability of PARP to repair single-strand DNA breaks results in double-strand breaks (DSB) and subsequent replication fork collapse, which can lead to cell death. DSB can be repaired via different mechanisms, including the predominate means homologous recombination (HR) repair, which depends on BRCA1 and BRCA2 functionality.10 Recent evidence has shown that PARP inhibitors (e.g., olaparib and talazoparib) are effective in treatment of malignancies that harbour deleterious gBRCAm compared to BRCA wild-type tumours (e.g., metastatic BC).11,12
The objective of this mini-review is to present the mechanisms of action of PARP inhibitors and briefly summarise the clinical trials leading to the approval of olaparib, talazoparib, rucaparib, and niraparib for treatment of BRCA-mutated, human epidermal growth factor receptor 2 (HER2)-negative metastatic BC. It should be noted that PARP inhibitors (which are well tolerated oral agents) are providing some reasonable hope for better outcomes and quality of life in a vulnerable population of patients with metastatic BC. In addition, this article discusses the efficacy, safety, and resistance to PARP inhibitors, focussing on women with metastatic BC.
The Role of Poly(ADP-ribose) Polymerase Enzymes in DNA Repair and the Insights into Targeted Synthetic Lethality of the Tumour
To ensure genomic integrity, cells can apply different mechanisms that identify and repair DNA injuries caused by exogenous or endogenous factors via highly coordinated DNA damage response systems.13 Several ‘sensors’ of DNA lesions are located in the cell nucleus, which communicate with effectors at the different cell sites.14 In particular, PARP1 (a member of the superfamily of ADP-ribosyl transferases that transfer poly[ADP-ribose] [PAR] or mono-ADP-ribose) is one such nuclear protein that is activated by DNA breaks.15 PARP1 is able to synthesise PAR chains, which are signals for the mobilisation of DNA repair proteins.16 Similarly, PARP2 and PARP3 display DNA-dependent (ADP-ribose) transferase activity.16 Poly-ADP-ribosylation (PARylation) relates to the covalent binding of negatively charged PAR on target proteins.16,17 It should be underscored that PARylation may destabilise or stabilise protein-DNA interactions, activate target proteins, and induce protein degradation by the proteasome.17 In addition, PARP proteins can regulate various cellular functions (e.g., DNA transcription and DNA damage response) via PARylation.15-17
At different points of the cell cycle, cells can apply several mechanisms for DNA repair. For instance, if single-stranded break repair is blocked and that cell tries to divide, the break can become double-stranded. At this point, if the cell has HR deficiency (HRD) it cannot survive. Moreover, cells that do not have BRCA1 or BRCA2 proteins (resultant of deleterious BRCA mutations) are very sensitive to PARP inhibition because they are unable to repair the DSB. This leads to synthetic lethality and cellular apoptosis.10,13 The concept of synthetic lethality has initiated the development of an innovative, genomically targeted therapy for patients with cancers that harbour gBRCAm (e.g., metastatic BC and OC).11,14 This therapeutic strategy is possible because the genomic instability of some cancer cells allows a novel class of medications, PARP inhibitors, to act specifically on tumour cells and spare healthy cells. In fact, many tumour cells with HRD are more susceptible to PARP inhibitors because of the fact that if DNA DSB are formed, and HR is the predominant repair mechanism in those cells, the unrepaired breaks are lethal. As a consequence, the patients with tumours harbouring deleterious BRCA mutations (e.g., germline or somatic, resulting in defective DSB repair by HR) can achieve the greatest
clinical benefits.13,16
Poly(ADP-ribose) Polymerase Inhibitors: An Overview of the Main Clinical Trials Leading to their Approval in the Metastatic Breast Cancer Setting
Recently, the following PARP inhibitors have received approval by the U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA): olaparib, talazoparib, rucaparib, and niraparib (Table 1).11,12,18-24
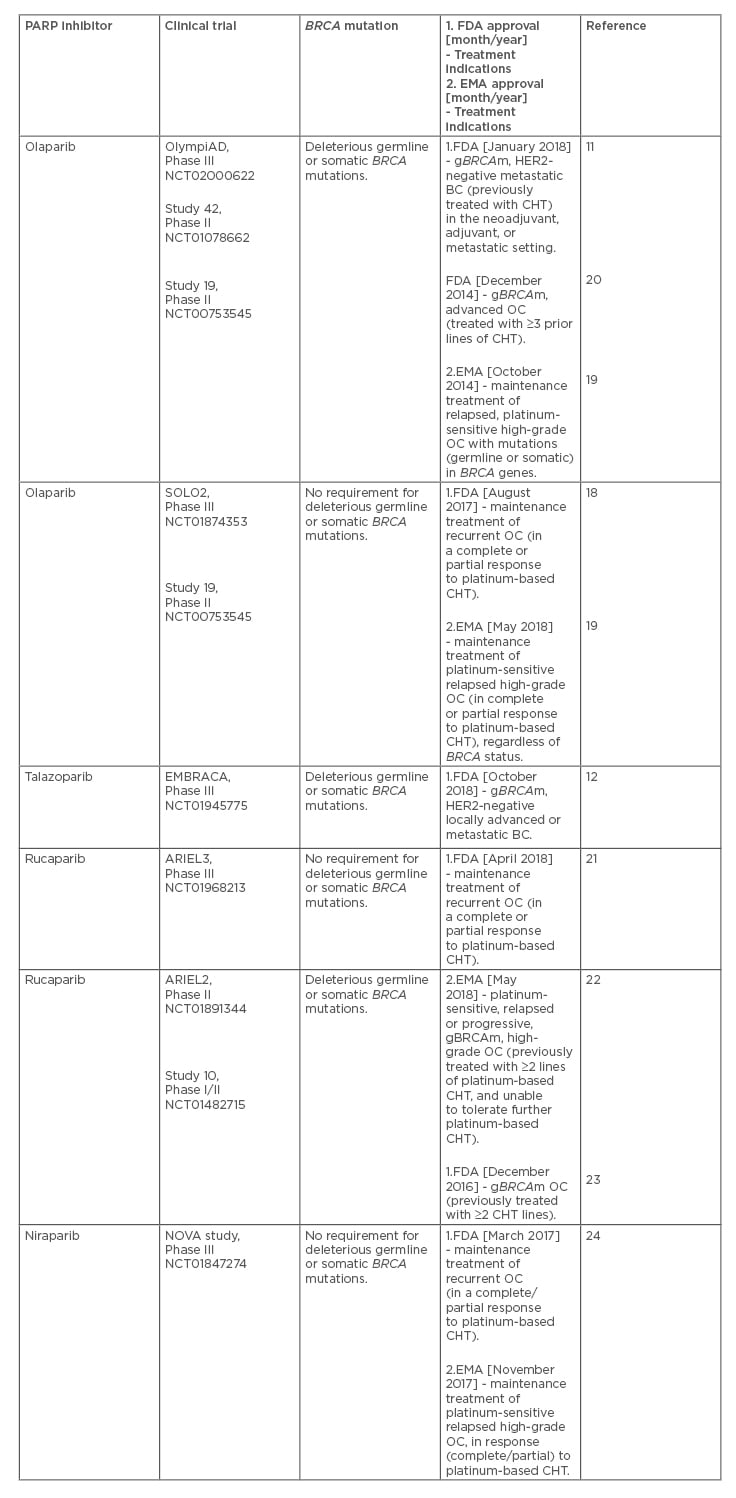
Table 1: Poly(ADP-ribose) polymerase inhibitors for the treatment of patients with advanced breast and ovarian cancer (based on the main clinical trials leading to their approval).
Olaparib was the first PARP inhibitor approved for the treatment of BC, based on the OlympiAD, Phase III randomised controlled trial, which has demonstrated that PARP inhibition was superior in the metastatic setting (e.g., third-line therapy), in terms of efficacy and safety, to chemotherapy (CHT).11 The CHT choices in OlympiAD included capecitabine, eribulin, and vinorelbine.11 As a consequence, olaparib has been indicated for deleterious or suspected deleterious gBRCAm, in women with HER2-negative metastatic BC, who have been treated with CHT (e.g., in the neoadjuvant, adjuvant, or metastatic setting).11 In addition, olaparib has been approved as maintenance therapy in patients with platinum-sensitive high-grade OC (with or without BRCA mutations)
(Table 1).18,19 Likewise, talazoparib has been approved for the treatment of women with deleterious gBRCAm, HER2-negative locally advanced, or metastatic BC (based on the EMBRACA, Phase III randomised control trial).12 From a clinical point of view, the findings of the OlympiAD and EMBRACA trials appear encouraging; however, it should be pointed out that the efficacy of PARP inhibitors was not compared to that of platinum CHT, and thus, the OlympiAD and EMBRACA studies could not evaluate the relative benefits of PARP inhibitors and platinum-based CHT in BC patients
with gBRCAm.11,12
Associations Between Germline BRCA Mutations and Triple-Negative Breast Cancer
BRCA1 and BRCA2 play an important role in repairing DNA injuries.25 It should be highlighted that the BRCA1 mutations are strongly associated with triple-negative breast cancer (TNBC) (oestrogen receptor-negative, progesterone receptor-negative, and HER2-negative BC).26 Conversely, the BRCA2 mutations are mostly associated with
hormone receptor-positive/HER2-negative BC.26 TNBC is the predominant subtype in women with a gBRCAm.26,27 However, the majority of basal-like BC are not in the BRCA1 carriers group, and 20% of genomic instability in TNBC can be explained by BRCA1 or BRCA2 inactivation.28 For instance, 5–10% of BC, and 80% of BRCA1-related BC, are TNBC.26,27 It should be noted that in TNBC, the BRCAness phenotype can be related to BRCA mutations, BRCA1 promoter methylation, or low BRCA1 mRNA or protein expression.27 Most BRCA1 carriers have basal-like BC. Among patients with TNBC, 14.6% have germline DNA repair gene mutations (e.g., 8.5% in BRCA1, 2.7% in BRCA2, and 3.7% in the PALB2 [partner and localiser of BRCA2], BARD1 [BRCA1 associated RING domain 1], RAD51 [RAD51 recombinase], or BRIP1 [(BRCA1 interacting protein
C-terminal helicase 1]).25,28
In a recent clinical trial, conducted among women with metastatic TNBC (with abnormal changes in DNA repair that were similar to those of BRCA-mutated tumours), 50% of the patients were treated with carboplatin, and the other 50% with docetaxel.29 It was shown in this unselected study population, that carboplatin and docetaxel revealed similar efficacy. It should be underscored that in women with gBRCAm, carboplatin doubled the response rate compared to those from the docetaxel group (68% versus 33%). However, this clinical advantage was not reported in patients with BRCA1 mRNA-low tumours, BRCA1 methylation, or HRD.29 It should be noted that with regard to olaparib, the reported response rate of 68% and the median progression-free survival (PFS) of 6.8 months were similar to those observed with carboplatin in metastatic TNBC.11 Since platinum sensitivity is associated with tumour susceptibility to PARP inhibitors, hopefully future large scale trials will compare the platinum-based CHT with the PARP inhibitors and show the application of carboplatin
in TNBC.
Resistance to Poly(ADP-ribose) Polymerase Inhibitors: Challenges and Considerations
PARP inhibitor resistance can develop via multiple mechanisms because BRCA1/2-deficient tumour cells can restore HR repair and stabilise their replication forks.30 In addition, it should be noted that there is some cross-resistance between platinum-based CHT and PARP inhibitors; however, this cross-resistance is incomplete, and it is still unclear to what degree the platinum compounds and the PARP inhibitors ‘operate’ on the same pathway. For instance, a study in women with OC has shown that patients who have platinum-sensitive cancers have also higher overall response rates to single-agent PARP inhibition. Conversely, patients with platinum-resistant or refractory tumours have revealed lower response rates.31 Unfortunately, not all patients with BRCA mutations are responsive to PARP inhibitors, indicating a potential role of the primary resistance to such a therapy. This may be attributable to the possibility that certain changes in the BRCA genes could have different functional influences on the individual response to PARP inhibitors. Moreover, an analysis of tumour biopsies has revealed some molecular mechanisms that can be ‘in charge’ of the primary and acquired resistance to PARP inhibitors. The most common acquired resistance mechanisms to PARP inhibitors consist of secondary mutations restoring the BRCA1 or BRCA2 protein functionality. Such mechanisms have been studied in patients with OC and BC (i.e., metastatic TNBC), who were resistant to platinum compounds.32,33
In a recent study, secondary somatic mutations that restored the open reading frame of BRCA or HR-related genes (e.g., RAD51C and RAD51D) were identified in patients with OC, who progressed during treatment with rucaparib.34 Furthermore, in patients with BRCA somatic heterozygous disruption, tumour progression was related to a recovery of BRCA activity. In contrast, patients with a single-copy loss of chromosome 17 and a somatic nonsense BRCA1 mutation (in the remaining allele) had a long-term response (>7 years) to olaparib. In this case, deletion of the wild-type allele resulted in the restoration to a functional gene.35 Some other clinically relevant resistance mechanisms can involve mutations or downregulation of PARP enzymes.36 It should be highlighted that HR gene sequencing, HR deficiency, genomic loss of heterozygosity tests, or some genetics scoring systems can allow detection of several patients as possible candidates for therapy with PARP inhibitors. However, such testing may still overlook some patients for whom the treatment with PARP inhibitors might be beneficial. Therefore, in the future, clinical studies should integrate all the data derived from DNA sequence and gene copy number variation that are relevant to other DNA repair mechanisms, such as nonhomologous end joining, alternative-nonhomologous end joining, and DNA damage regulatory gene processes.34 In the meantime, gBRCAm can be considered as an indicator for targeted therapy with
PARP inhibitors.
Poly(ADP-ribose) Polymerase Inhibitors: Clinical Implications, Concerns, and Future Directions
PARP inhibitors are generally well tolerated, and their adverse effects (AE) are mostly related to haematologic and digestive tract toxicity (Table 2).37,38
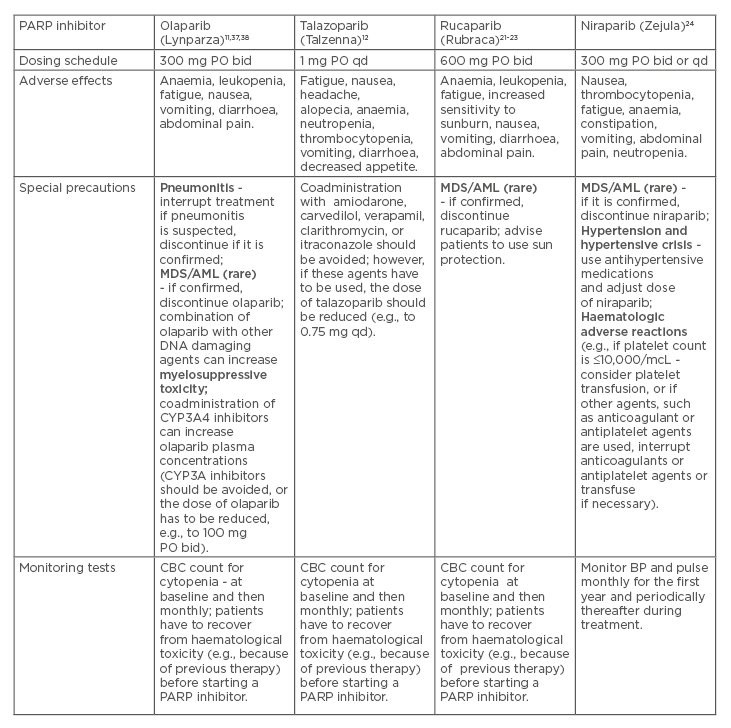
Table 2: Poly(ADP-ribose) polymerase inhibitors: Dosing schedules, adverse effects, and special precautions.
AML: acute myeloid leukaemia; bid: twice a day; BP: blood pressure; CBC: complete blood count; CHT: chemotherapy; ET: endocrine therapy; HER2: human epidermal growth factor receptor 2; HR: hormone receptor; MDS: myelodysplastic syndrome; PARP: poly (ADP-ribose) polymerase; PO: per os (orally); qd: once a day.
Such AE can be successfully managed by adjusting doses, and using symptomatic medications or transfusions, if necessary.37,38 Although PARP inhibitors have revealed therapeutic efficacy mostly in women with advanced BC who harbour gBRCAm, their effectiveness outside of gBRCAm carriers (e.g., in case of somatic BRCA mutations or other genetic mutations) is an important area of future studies. Furthermore, novel predictive biomarkers of responsiveness (beyond gBRCAm) are needed in patients with metastatic BC. In addition, exploring the use of PARP inhibitors in combination with other anticancer therapies presents an ongoing challenge. In practice, the PARP inhibitors can be used as single agents, according to the concept of synthetic lethality (because of the defects in HR).39 Moreover, the PARP inhibitors can be used in combination with other treatments (e.g., CHT or radiotherapy) because PARP inhibitors augment DNA damage and contribute to an increase in overall DNA damage in tumour cells, even without the presence of HRD.39
Olaparib, applied as a single agent, has been beneficial in patients with advanced BC and OC, and the median PFS in the case of BC is approximately 6 months.33 Similarly, talazoparib, which is the most potent PARP inhibitor, has been used as a single agent in women with BC harbouring gBRCAm, in which the median PFS was almost 9 months.40 It should be underscored that for patients with advanced and metastatic BC, BRCA testing is very important, together with possible testing for other germline mutations since PARP inhibition is an effective targeted therapy for gBRCAm. However, some important questions, which will hopefully be answered in the future trials, involve the following issues:
Which therapies are most optimal for combination with PARP inhibitors?
Should PARP inhibitors be introduced into the early-stage BC treatment?
How effective are PARP inhibitors in BC patients with prior exposure to platinum-based CHT?
Can intermittent therapeutic dosing of PARP inhibitors be used?
Conclusion
For many patients with metastatic BC, treatment with PARP inhibitors can be more effective and less toxic than that of CHT. Because of the underlying defects in DNA repair, cancer cells with BRCA mutations are vulnerable to therapies that target PARP. In particular, for malignant tumours with HRD, PARP inhibitors induce synthetic lethality.
Some subtypes of BC with gBRCAm or functional (nonmutational) defects in BRCA proteins represent therapeutic targets for
PARP inhibitors. Clinical benefits of PARP inhibitors have mostly been accomplished in BRCA-associated cancers (such as advanced BC and OC). BC with defects in a specific DNA damage repair pathway is particularly sensitive to targeted therapy with PARP inhibitors. For instance, olaparib (the first PARP inhibitor approved by the FDA) has currently been indicated for the treatment of patients with HER2-negative metastatic BC previously treated with CHT and who harbour deleterious gBRCAm. The recent OlympiAD trial has revealed a significant PFS benefit of olaparib compared with the CHT. In addition, PARP inhibitors may offer an effective and safe therapeutic option for women with TNBC. The most common AE of PARP inhibitors include anaemia, neutropenia, nausea, and vomiting.
It should be highlighted that identifying possible predictors of response to PARP inhibitors and strategies to overcome various mechanisms of resistance merit investigation in the future clinical trials. In particular, the HRD assays attempt to use chromosomal instability as a marker. Also, there are many candidate genes beyond BRCA1/2 that are involved in HR (e.g., RAD51D). Advances in genome-sequencing of tumour DNA, combined with modern bioinformatics, will hopefully contribute to defining a more precise panel of genes that will be helpful in determining profiles of individual patients who may favourably respond to the therapy with PARP inhibitors.






