
Abstract
Radiotherapy is one of the most conventional modes of treatment in several cancers. Failure of radiotherapy followed by acquisition of radioresistance is one of the emerging challenges faced by clinical experts. Unusual expression and functional implications of several molecules are observed to facilitate radioresistance. Aurora A, a member of the Aurora kinase (serine/threonine kinase) family, is one such molecule that shows significantly altered expression as well as non-canonical functional crosstalk with other associated factors (cell cycle regulators, signaling molecules, stemness markers, etc.) to favour the adaptations for the acquirement of radioresistance. These mechanisms include progression of cell cycle, stimulatory activation of factors by phosphorylation for enhancing the chance of cellular survivability, and prevention of apoptosis. This review article summarises how Aurora A is responsible for radioresistance in cancer and why this kinase should be considered a negative biomarker of radiosensitivity. This review discloses a wider opportunity in the field of research to find the mechanistic key regulatory pathway of Aurora A, which can be a potential target for enhancing the efficiency of treatment. Further investigations are required to explore the potential of Aurora A inhibitors as reliable radiosensitisers.
INTRODUCTION
Radioresistance is known to create complications in the treatment of cancer.1 Radiation-induced altered adaptive responses by tumour cells or tissues are considered to be primary reasons behind the failure of radiotherapy.2 Acquirement of radioresistance followed by treatment failure and locoregional recurrence is a multifaceted process. Emerging data have suggested various molecular biomarkers necessitate radioresistance in cancer.3,4 It is well established that the level of radioresistance alters in different phases of the cell cycle. Several researchers have experimentally proven that most cells are resistant in the G0 phase, early G1 phase, and late S phase of the cell cycle. In contrast, most cells are radiosensitive in the late G1 phase, G2 phase, and throughout the M phase of the cell cycle.5 In the S phase, radiation resistance is thought to be due to an increased amount of DNA synthesis and repair enzymes, as well as a rise in the intracellular levels of glutathione (a free radical scavenger). In response to ionising radiation, the G1 phase of the cell cycle is generally blocked to allow time for the recognition and repair of DNA damage prior to the initiation of DNA synthesis. Cells show the most sensitivity towards radiation during the G2/M phase of the cell cycle because of the lack of time for adequate repair before chromosome segregation. The key participating genes and their products halting cell cycle progression, which increase or are post-translationally altered following DNA damage, are p53, p21, growth arrest and DNA damage-inducible protein 45, and the retinoblastoma protein. Overall, there is a damage-responsive G1 block, halting the cell cycle at G2/M until DNA damage can be repaired. Thus, those cells having the ability to repair the damaged DNA may improve cell survival and impart additional resistance towards radiation.6,7 It is widely reported that the essential mechanism by which ionising radiation exerts its therapeutic effect is by induction of DNA damages, such as double-strand breaks, single-strand breaks, and oxidation of DNA bases. These damages may be removed by homologous recombination, non-homologous end joining, single-strand break repair, and base excision repair.8 Now, it is of importance to note why and how these participating genes get altered following DNA damage. Aurora A is found to interact with most of these regulators of cell cycle checkpoints to override their effect and continue cell cycle progression, and thereby contributes to radioresistance.
Three families of human Aurora kinases, Aurora kinase A, B, and C, share a common C-terminal catalytic domain in their proteins.9 These three kinases are known to participate in mitotic progression of cells; however, little is known about Aurora kinase C.9 During mitosis, centrosome maturation and separation followed by the assembly and stability of spindle is controlled by Aurora Kinase A.10 Aurora kinase B is a member of the chromosomal passenger complex along with other members (survivin, borealin, and INCENP).11 This kinase allows proper segregation of chromosomes and completes the cytokinesis process.11 Evidence suggests that the zone of Aurora kinase B localisation is at the K-fibres, which are the specialised microtubules near kinetochores;11 however, this localisation varies in different phases of the cell cycle, and is at the chromosomes in prophase, the centromere in prometaphase and metaphase, and the central mitotic spindle in anaphase.12 Although Aurora kinase A and B are found in most of the somatic cells, Aurora kinase C is distributed in a limited manner to germline cells (sperm and oocyte) undergoing meiosis.13 Aurora C primarily acts as a regulator of chromosome segregation in a lesser-known mechanism. The cell cycle specific distributions of these kinases are summarised in Figure 1.
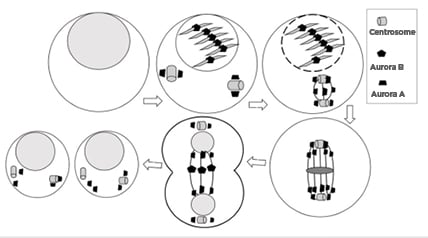
Figure 1: Cell cycle phase-specific distribution of Aurora kinases.
Aurora A localises to the centrosome from G1/S phase, with the progression of cell cycle it localises in spindle poles until telophase. Aurora B localises to the kinetochore followed by midbody of the central spindle during mitosis. The progression of cell division is shown using arrow signs from left showing normal cell, cover by onell in early prophase, cell in late prophase, cell in metaphase, cell in anaphase, and cells after division.
Apart from the canonical activities regulated by Aurora A, its overexpression also correlates with acquired radioresistance in several cancers. This review highlights some of the mechanisms performed by Aurora A in imparting radioresistance in cancer.
AURORA A
Salient Structural Features
Aurora A (or STK15/BTAK) is a well-known cell cycle regulatory kinase. Being a mitotic kinase, it is a member of the serine/threonine protein kinase family, which plays an important role in proper entry of cells in mitosis, formation of a bipolar spindle, control of centrosome maturation, and appropriate chromosomal segregation during mitosis.8 If the activity of Aurora A is suppressed by RNA interference, it results in delayed mitotic entry of cells,14 whereas centrosome amplification, cytokinesis inhibition, and aneuploidy are some well-verified after-effects of its overexpression.15 Before looking into the functional mechanism of Aurora A, it is essential to understand the different protein domains of this kinase. The Aurora A gene has its chromosomal location at 20q13.2, which is often found to be significantly amplified in several human epithelial tumours.16 The molecular weight of the mammalian Aurora A protein is 46 kDa and the protein is 402–403 amino acids long. The common structural configuration of all Aurora kinases includes an N-terminal domain (39–139 amino acids), a kinase domain (250–300 amino acids), and a C-terminal domain (15–20 amino acids).17 The kinase domain of Aurora A is mainly composed of a β-stranded N-terminal lobe and α-helical C-terminal lobe that are linked together by a hinge region in order to acquire the active conformation.18 The ATP-binding domain of Aurora A consists of three specific sequence variants (leucine 215, threonine 217, and arginine 220), of which the threonine 217 locus particularly distinguishes Aurora A from Aurora B kinase domains.19 These particularities of the Aurora A domain are useful for the design of specific Aurora A inhibitors. Some non-catalytic domains present in Aurora A trigger its degradation. These include the D-boxes, KEN motifs, and the DAD/A boxes. Mutation of the C-terminal D-box sequence is essential for the stability of Aurora A, and degradation of Aurora A depends on D-Box instead of KEN-box motifs (residues 6–9). Aurora A degradation is also mediated by the anaphase promoting complex or cyclosome, which occurs at an atypical degradation sequence present in Aurora A named the DAD/A box.20,21 Aurora A possesses a short α-helix called the B-helix, located just prior and perpendicular to the C-helix. The structure of Aurora A is depicted in Figure 2.
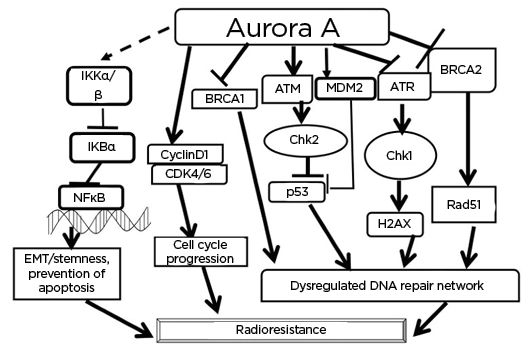
Figure 2: Some of the well-studied interactions of Aurora A that are found to help in acquirement of radioresistance.
Aurora A activates NFκB and generates EMT and causes prevention of apoptosis. The kinase can activate cyclin D1 and CDK to allow progression of cell cycle. Aurora A, by interacting with DNA repair regulators (BRCA1, BRCA2, ATM and Rad3-related kinase, Chk 1/2, and p53), causes dysregulated DNA repair. All of these result in changes that lead to radioresistance.
ATM: ataxia–telangiectasia-mutated kinase; ATR: ataxia–telangiectasia-mutated and Rad3-related kinase; BRCA: breast cancer susceptibility protein; CDK4/6: cyclin-dependent kinase 4/6; Chk: checkpoint kinase; EMT: epithelial–mesenchymal transition; MDM2: mouse double minute 2 homolog.
The C-terminal region of the kinase domain consists of seven α-helices and a two-stranded β-sheet, and contains the catalytic aspartic acid of the HRD motif (sequence histidine, arginine, and aspartate at positions 254–256) and the mobile activation loop, whose position and conformation determine whether a kinase is active or inactive.22,23 The Aurora A activation loop (the most crucial part required for functional activation of the protein) spans 274–299 amino acid residues, beginning with an aspartate, phenylalanine, and glycine sequence motif and ending with an alanine, proline, and glutamate motif (sequence proline, proline, and glutamate in Aurora A). Aurora A becomes active by its autophosphorylation (discussed in detail later) and trans-phosphorylation at threonine 287 and threonine 288 residues by targeting protein for Xenopus kinesin-like protein 2,24 p21 activated kinase,25 protein kinase A,26 or atypical protein kinase C.27
Localisation
Being a centrosomal protein, Aurora A resides next to the centrosome late in the G1 phase and early in the S phase. The N-terminal domain of Aurora A participates in the localisation of the kinase to the centrosome during interphase. With the progression of the cell cycle, Aurora A concentrations increase and an association of the kinase with the mitotic poles and the adjacent spindle microtubules is observed. Such association lasts until telophase. Re-localisation of Aurora A to the mid-zone of the spindle occurs immediately before mitotic exit.28
AURORA A ACTIVATION
Transcriptional
The cell cycle-induced transcription of Aurora proteins is made possible by the presence of the cell cycle-dependent element and cell cycle gene homology region in Aurora A promoters.
The cell cycle-dependent element and cell cycle gene homology region sequences in Aurora A mediate the transcription of Aurora A and other crucial G2/M regulators (e.g., cyclin A, cell division cycle 25 phosphatase, cyclin-dependent kinase [CDK] 1, and polo-like kinase).29
Post-translational Modifications
Post-translational modification is an important prerequisite for functional activation of the enzyme. Like other serine/threonine kinases, functional regulation of Aurora A occurs through activation loop phosphorylation. Aurora A auto-phosphorylation, a salient post-translational modification, is mediated by several co-factors, of which Ajuba, targeting protein for Xenopus kinesin-like protein 2, protein aurora borealis, and transforming acidic coiled–coil-containing protein 3 are the noteworthy ones. As described earlier, Aurora A has two regulatory sites for phosphorylation in its activation loop (threonine 287 and threonine 288). Phosphorylation of Aurora A in its catalytic domain at threonine 288 is known to trigger the kinase activity of the enzyme.24 This phosphorylation eventually causes a positive feedback loop, which is responsible for maintaining the activated state of the kinase until anaphase. Maximum activity has been observed from late G2 until pro-metaphase. The function of threonine 287 phosphorylation is still unclear.30
RADIORESISTANCE IN CANCER: FUNCTIONAL ROLE OF AURORA A
Apart from its intricate role in regulating mitosis, activated Aurora A also subsequently promotes a variety of biological functions for maintaining cancer phenotypes. Some of the functions of Aurora A include cell proliferation, migration, invasion, epithelial–mesenchymal transition, and maintenance of cancer stem cell behaviors.31 Now, it is necessary to understand whether Aurora A has any impact on radiotherapy in cancer. Ectopic expression of Aurora A led to increased sensitivity to ionising radiation in MCF10A normal breast epithelial cells.30 In contrast, in cancer cells, a plethora of evidence suggests that excessive Aurora A attenuates the radiosensitivity of cells, while silencing or inhibition of Aurora A with small interfering RNA or selective inhibitors enhances radiosensitivity.32 The possibility of involvement of Aurora A in contributing radioresistance has been experimentally tested in several cancers. Overexpression of Aurora A in cervical squamous cell carcinoma showed a strong positive correlation with cancer cell invasion and metastasis. Furthermore, Aurora A is considered to serve as an independent prognostic factor, which affects disease-free survival and overall survival.33 In one study, patients with cervical cancer were treated with Aurora A inhibitors. Treatment outcomes included increased cell apoptosis after X-irradiation and downregulation of the expression of cyclin D1, CDK2, and CDK6, which eventually induced cell cycle arrest at the G2/M phases.34 These findings indicate that Aurora A activation gives rise to radiation resistance by allowing cell cycle progression in cervical cancer. Therefore, Aurora A may be regarded as one of the therapeutic targets and an independent prognostic factor for increasing the sensitivity of cervical squamous cell carcinoma radiotherapy. A predominant finding of Shen et al., who studied hepatocellular carcinoma (HCC) cell lines, revealed lower expression of nuclear IκBα protein in parental HCC cells at the same time as higher expression levels of p65 protein in radioresistant counterparts. Knockdown of Aurora A led to increased expression of nuclear IκBα proteins by way of decreased expression of p65 proteins in radioresistant HCC cells. The expression of downstream effectors of the NF-κB pathway, such as B-cell lymphoma 2, myeloid cell leukaemia 1, cleaved poly(ADP-ribose) polymerase, and caspase-3 (cysteine-aspartic proteases), is enhanced by hyperproduction of Aurora A. Triggering apoptosis in radioresistant cells was attained by knockdown of Aurora A, which resulted in downregulated B-cell lymphoma 2 and myeloid leukaemia 1 protein expressions as well as upregulated cleaved poly(ADP-ribose) polymerase and caspase-3 expressions in radioresistance.35 To explore whether Aurora A contributes to radioresistance, a study was conducted by Sun et al., using Aurora A complementary DNA/short hairpin RNA or the specific inhibitor VX-680 (Vertex Pharmaceuticals, Inc., Boston Massachusetts, USA). This study confirmed that Aurora A positively regulates cell proliferation, cell cycle progression, and anchorage-independent cell growth in order to establish resistance against X-rays. Simultaneous promotion of the expression of ataxia–telangiectasia-mutated kinase/checkpoint kinase 2 and suppression of the expression of breast cancer susceptibility protein 1/2, ataxia–telangiectasia-mutated and Rad3-related kinase/checkpoint kinase 1, p53, phospho-p53 (serine 15), H2AX, γH2AX (serine 139), and RAD51 by Aurora A resulted in a dysregulated DNA repair mechanism. The formation of a γH2AX focus, which is considered as one of the prognostic markers of radiation-induced DNA damage, was found to be minimised by Aurora A. These reports account for supportive evidence of the hypothesis that Aurora A is a negative biomarker of radiosensitivity.36 Similar results were observed in HeLa cells, which were irradiated with 4 Gy of γ-rays. The electrophoretic mobility shift assay, luciferase reporter gene assay, immunoblot analysis, small interfering RNA-based gene knockdown, and overexpression studies concluded that Aurora A enhances the binding of NF-κB to DNA, thereby increasing the gene transcription by NF-κB and decreasing the radiosensitivity of the cells.37 Equivalent results were obtained in radioresistance of lung adenocarcinoma.38 The effectiveness of radiation-induced cell death often relies on the mechanism of reactive oxygen species-mediated cellular damage. Tumour tissues that are deprived of adequate oxygen supply due to hypoxic conditions lack sufficient amounts of reactive oxygen species, thereby causing a three-fold increase in the chance of acquired radioresistance. The transformed tumor cells get adapted in the hypoxic tumour microenvironment by developing resistant characters in them.39 Simultaneous overexpression of hypoxia-inducible factor-1α and Aurora A is a remarkable event during hypoxia, which establishes a positive feedback between these two molecules.40 It has been suggested that hypoxic cellular proliferation and migration are clinically correlated with increased mRNA and protein expression of Aurora A. Conversely, inhibition of Aurora A could reverse this event.40 In a further study, it was confirmed that in hypoxia, hypoxia-inducible factor-1α binds at the promoter region of Aurora A and enhances its transcription.41 This associative involvement of Aurora A in both hypoxia and radioresistance is notable, which again provokes the possibility of this kinase in imparting radioadaptive responses during acquirement of radioresistance. Experimental evidence indicated that MLN8237 (Takeda Pharmaceutical Company Ltd., Tokyo, Japan), a selective Aurora A inhibitor, could induce cell cycle arrest at the G2/M phase and significantly reduce radiation-dependent resistance.42 Similar studies were found in selected lung cancer cell lines, where inhibition of Aurora A enhanced radiosensitivity.43 Increased expression of Aurora A is negatively correlated with survival in patients with non-small cell lung carcinoma.43 One of the established Aurora kinase inhibitors, daurinol, targets the kinase and enhances radiotherapy;44 however, the exact mechanism concerning the functional impact of this Aurora A inhibitor needs to be investigated thoroughly. Whether the inhibitor enhances radiosensitivity by directly blocking Aurora A or through inhibiting cyclin expressions (cyclin D1, CDK2, and CDK6) is yet to be elucidated.44 Aurora kinase expression was reported to be regulated transcriptionally by radiation because the mRNA and protein expression of such kinases were increased by sub-lethal doses of radiation.45 The level of Aurora kinases present in pre-treatment biopsies could serve as a predictive factor to identify patients likely to respond to conservative radiotherapies. Pharmacological approaches targeting Aurora kinases in tumors over-expressing these proteins could strongly increase the therapeutic ratio in radiotherapy for cancer treatment. The underlying mechanisms by which targeting aurora kinases may improve the response to radiation seem to be multifaceted and involves cell cycle distribution.43 In radioresistant pancreatic cancer cells, treatment with an Aurora A inhibitor resulted in co-inhibition of cyclin D1, CDK2, and CDK6 to induce cell cycle arrest at the G1/S and G2/M phases, and also promoted cell apoptosis after γ-irradiation.46 In laryngeal cancer cells, inhibiting Aurora A by VX-680 induced expression of p53 and potently sensitised cells to radiotherapy, leading to significant cell death, whereas ectopic overexpression of Aurora A reduced p53 levels and rendered cells more resistant to irradiation. Taken together, Aurora A kinase, a negative prognostic marker, promotes migration, and reduces radiosensitivity.47 The Aurora A signaling axis pertinent to radioresistance is represented in Figure 2.
OTHER PROBABLE INTERACTIONS MEDIATED BY AURORA A IN RADIORESISTANCE
Radioresistance is a collective contribution of several factors, with which Aurora A interacts. Signaling pathways like phosphatidylinositol 3-kinase/protein kinase B, NF-κB, and Wnt/β-catenin in association with stemness markers like sex-determining region Y-box 2, octamer-binding transcription factor 4, and Twist1 guide the cells to obtain epithelial–mesenchymal transition-like characteristics, which accounts for stabilisation of the resistant phenotype.48-59 Aurora A is found to interact with these molecules during cancer progression. Therefore, there are strong possibilities that such interactions play an essential role in Aurora A-mediated radioresistance. Some of these interactions are summarised in Table 1.
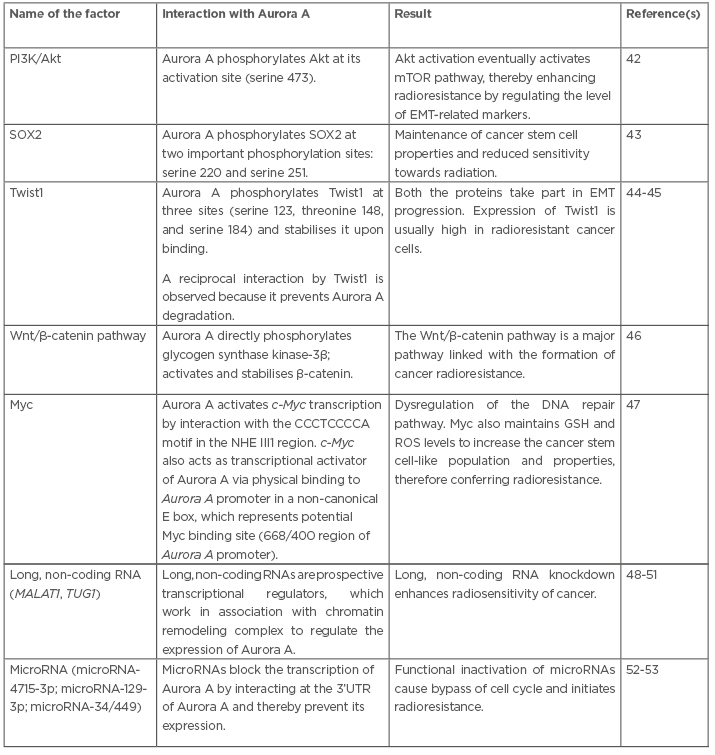
Table 1: Interactions of several factors with Aurora A that have a potential to generate radioresistance.
Akt: protein kinase B; EMT: epithelial–mesenchymal transition; GSH: glutathione; MALAT1: metastasis-associated lung adenocarcinoma transcript 1; mTOR: mechanistic target of rapamycin; NHE III1: nuclease hypersensitive element III1 region; PI3K: phosphatidylinositol 3-kinase; ROS: reactive oxygen species; SOX2: sex determining region Y-box 2; TUG1: taurine up-regulated 1; 3’UTR: three prime untranslated region.
CONCLUSIONS AND PERSPECTIVES
One of the major reasons behind the therapeutic failure of radiation is radioresistance. Cancer cells that survive the effects of radiation acquire adaptations to bypass cell cycle checkpoints and thereby retain their proliferative and invasive properties. Several faulty repair mechanisms accumulate changes in sequences of DNA, which makes this process smoother. The regulatory role of Aurora A in mitotic and non-mitotic events involves several molecules, ranging from DNA repair mediators to signaling modulators. It is more important to highlight that blocking and inactivation of Aurora A activity can reverse the process of radioresistance and increase radiosensitivity. Treatment of patients using fractionated irradiation determines acquirement of resistance against radiotherapy, which is clinically complemented with overexpression of Aurora A. Thereby, Aurora A can serve as a prognostic marker of radioresistance. This review provides a thought-provoking element to global researchers, encouraging them to design novel experiments in order to explore the mechanistic key regulatory pathway of Aurora A necessary for enhancing the efficiency of treatment. Checking the levels of Aurora A along with administration of Aurora A inhibitors prior to radiotherapy could be applied in the future to enhance the efficacy of radiotherapy. Clinical administration of Aurora A inhibitors as reliable radiosensitisers may serve as a scope of future investigations. The details of several Aurora kinase inhibitors are provided below.
Type of Inhibitor
Pan-aurora inhibitor
Tozasertib (VX-680/MK0457 [Merck & Co., Kenilworth, New Jersey, USA]) is currently in Phase II clinical trials, and exerts its activity by inducing apoptosis and autophagy.60 Danusertib (PHA-739358), which is also in Phase II clinical trials, blocks the activities of fibroblast growth factor receptor 1, Abl, rearranged during transfection, and tropomyosin receptor kinase A, and increases expression of the p53 protein and its downstream effector protein p21.61
PHA-680632, CYC-116, SNS-314, R763, and AMG-900 are currently in Phase I clinical trials. PHA-680632 shows additive effects in cancer cells in association with radiation. Treatment with PHA-680632 prior to ionising radiation causes enhancement in apoptosis, micronuclei formation, and breast cancer susceptibility protein 1 foci formation.62 CYC-116 inhibits Aurora activity by interfering with its autophosphorylation, reducing histone H3 phosphorylation and subsequent polyploidy, and ultimately causing failure in cytokinesis followed by cell death.63 SNS-314 exhibits potent and sustained responses, including reduced phosphorylated histone H3 levels, increased caspase-3, and appearance of increased nuclear size.64 R763 causes enlargement of cell size, endoreduplication, and apoptosis.65 AMG-900 is an ATP-competitive phthalazinamine small molecule inhibitor of Aurora kinases. AMG 900 inhibits autophosphorylation of Aurora kinase and phosphorylation of histone H3 on serine 10. This leads to aborted cell division without a prolonged mitotic arrest, which ultimately results in cell death.66 AT-9283 is currently in Phase II clinical trials, and promotes a clear polyploid phenotype by inhibiting the activity of Aurora kinase.67 Finally, PF-03814375, which is in Phase I clinical trials, is a novel, potent, orally bioavailable, reversible inhibitor of Aurora kinase.68
Aurora A inhibitor
Alisertib (MLN8237), which is in Phase II clinical trials, performs cell cycle arrest at the G2/M phase, and instigates apoptosis and senescence. It allows up-regulation of p53, p21, and p27, and cleavage of poly(ADP-ribose) polymerase, caspase-3, and caspase-9.69 ENMD-2076 is also in Phase II clinical trials, and inhibits the activity of Aurora A and B kinases, as well as inducing G2/M cell cycle arrest.70 Lastly, MLN8054 is an ATP-competitive, reversible inhibitor of Aurora A kinase. Its mechanism of actions includes halting cell proliferation by promoting G2/M accumulation and spindle defects. A recent study showed that MLN8054 sensitises androgen-resistant prostate cancer to radiation by inhibiting Aurora A kinase, which is associated with sustained DNA double-strand breaks.71,72






