
INTRODUCTION
Mismatch repair-deficient (MMR-D) tumours exemplify the prototypic hypermutator phenotype. Owing to the high mutation rates, many neo-antigens are present on the tumour cells’ surface, typically shared among different cancer types. The MLH1 knockout mouse represents a preclinical model that resembles features of the human MMR-D counterpart.1,2 These mice develop MMR-D neoplasias spontaneously.
The tumour spectrum is complex, with a high prevalence of early non-Hodgkin T cell lymphomas (NHL), lymphoid skin lesions, and at later stages, epithelial tumours of the gastrointestinal tract (GIT). The sequential appearance of these neoplasias, often in a mutually exclusive fashion (the ‘either-or’ principle of tumour type), raises the question of how the biological and/or molecular mechanisms of the neoplasia relate to each other.
METHODS
Using whole-exome sequencing on MLH1-/- primary tumours (two GIT, one splenic NHL, and one skin lymphoma) as well as GIT-derived cell lines (n=2), the study aimed to identify underlying molecular alterations. The researchers focussed on shared and mutually exclusive mutations, and described the processes of ongoing mutational events in tumour-derived cultures. Finally, the coding microsatellite mutational profile was examined on a panel of primary tumours to detect shared mutations in MMR-D target genes.
RESULTS
MLH1-/- tumours show high tumour mutational burden with three of four primary tumour samples being ultra-hypermutated (>100 mutations/megabase). Missense mutations were more frequent than nonsense mutations, and base changes were mainly due to transitions (C>T; A>G). The resulting mutational landscape was heterogeneous and in accordance with the human counterpart; MLH1-/- tumours frequently harbour mutations in PIK3CA, EGFR, BRAF, KRAS, and ERBB3.1 Of note, only a few shared mutations were detectable among different tumour entities (ARID1A and IDH2). Mutations in the tumour suppressor genes SMAD4 and POLE were mutually exclusive in lymphomas, most likely contributing to a more aggressive in vivo phenotype (Figure 1). POLE mutations affect the exonuclease domain, similar to the human MMR-D counterpart.3,4 As a consequence, affected cells rapidly accumulate point mutations, yielding the ultrahypermutated phenotype. This was similar to the observations made in this study. Comparing the mutational profile of selected primary tumours, and their corresponding cell line, in vitro culture revealed continuously increased numbers of somatic gene mutations. The same was true for coding microsatellite mutations in selected MMR-D target genes, showing a gradual increase during in vitro passage. With respect to the latter mutation type, a partial overlap was detectable, and shared antigens were recognised. The two most promising candidates are AKT3, a RAC-gamma serine/threonine-protein kinase involved in the maintenance of cellular homeostasis, and ERCC5 (Excision Repair 5, Endonuclease), involved in DNA excision repair. Mutations in this gene increase susceptibility for skin cancer development. Given the biological function of these two genes, their applicability as a target is worth determining.
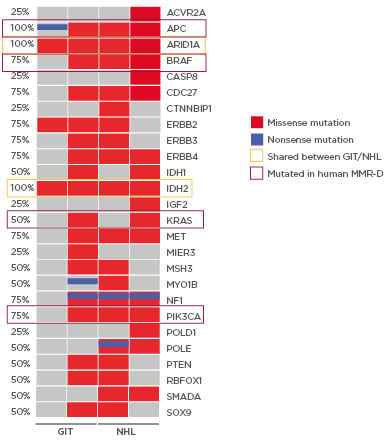
Figure 1: Mutational signature of MLH1-/--associated tumours in mice.
Oncoprint presenting genes with tumour suppressive function as well as relevance for tumour initiation and progression. Non-synonymous alterations are shown for particular genes (rows) affecting particular individual samples (columns). Shared mutations (orange box) as well as mutations conserved in the human counterpart (red box) are highlighted. Red bars indicate missense and blue bars nonsense mutations.
GIT: gastrointestinal tumour; NHL: non-Hodgkin lymphoma.
CONCLUSION
This pilot study is the first reporting results of a comparison between different spontaneously developing tumours as models for MMR-D driven tumourigenesis. ARID1A was identified as a potential secondary causative mutation hotspot, which is in line with the human counterpart associated with shortened time to cancer-specific mortality. This comprehensive characterisation of the mutational landscape may, therefore, be a good starting point to predict antigens for vaccination approaches.






