
Abstract
Seizures are one of the most common medical problems affecting children, and epilepsy is the most common chronic neurological condition in children. Childhood epilepsy syndromes include a wide spectrum of disorders ranging from benign to life threatening. While there are many known epilepsy syndromes, there are many factors, which may lead to the development of seizures in children including infection, traumatic brain injury, or structural abnormality.
Up to 40% of childhood epilepsies are thought to have some component of genetic involvement. New genes, mutations, and variants involved in epilepsy are being identified continuously. Most of the genes which have been identified encode for neurotransmitter receptors, ion channels, molecules involved in intracellular signalling, or proteins involved in synaptic structure. As new candidate genes in epilepsy are identified, new technologies in genetic testing are becoming available and more accessible, making the molecular diagnosis of epilepsy increasingly relevant to researchers, physicians, patients, and their families.
The standard of care and first-line treatment is the use of antiepileptic drugs. For those patients with medication-refractory epilepsy other available therapies include ketogenic diet, vagal nerve stimulator, or epilepsy surgery. The newest advancement in the treatment of paediatric epilepsies is based around the idea of targeted therapy. These therapies incorporate pharmacogenomics, the principle that an individual’s genetic background affects their response to specific drugs, as well as precision medicine, which identifies treatments for the damaged products resulting from specific gene mutations. Many of these therapies are still under research or in trial; however, there is much promise for the future of targeted medications.
INTRODUCTION
Seizures are one of the most common medical problems affecting children, and epilepsy is the most common chronic neurological condition in children. The incidence of epilepsy in children according to a 2012 cohort study is 144 per 100,000 person-years in the first year of life, and 58 per 100,000 person-years up to age 10.1 The cumulative incidence of childhood epilepsy was found to be 0.45% at the age of 5 and 0.66% at the age of 10.1 In 2005, it was found that children below the age of 10 have a lifetime prevalence of epilepsy of 0.6%, and that up to 5.0% of children will have a febrile seizure by the age of 5.2
In 2014, the International League Against Epilepsy (ILAE) redefined epilepsy as meeting one of three inclusion criteria: 1) at least two unprovoked seizures occurring >24 hours apart; 2) one unprovoked seizure and a probability of further seizures similar to the general recurrence risk (at least 60%) after two unprovoked seizures, occurring over the next 10 years; 3) the diagnosis of an epilepsy syndrome.3 Recent advances in genetic testing, as well as increased awareness surrounding childhood epilepsy, have enabled diagnoses to be given earlier and with more accuracy.
CHILDHOOD EPILEPSY SYNDROMES
Childhood epilepsy syndromes include a wide spectrum of disorders ranging from benign to life-threatening. Self-limited epilepsy with centrotemporal spikes accounts for 15–25% of all childhood epilepsies and is the most common idiopathic childhood epilepsy syndrome in children.4 Peak onset is between the ages of 7 and 8 years and usually resolves by age 16. It has the best prognosis of all epilepsy syndromes.5 Seizures typically present at night and are usually focal involving one side of the face, lips, or tongue.4 Recent studies have shown, that although self-limited epilepsy with centrotemporal spikes has a very favourable prognosis, some children may have long-term cognitive deficits, including language, memory, and auditory processing impairments.6
Self-limited occipital epilepsy is another childhood epilepsy syndrome and accounts for 5–10% of epilepsies.4,7 There are two types, early onset which is known as Panayiotopoulos syndrome and late onset, which is known as Gastaut type. In Panayiotopoulos syndrome, the age of onset is between the age of 1 and 6, and seizures typically resolve within 3 years. It presents as a triad of symptoms of nocturnal seizures, tonic eye deviation, and vomiting.7 Prognosis is very good and antiepileptic drugs (AED) are usually not needed. The Gastaut type begins later in childhood with peak incidence at 8 years old. It is characterised by brief seizures with visual symptoms, such as simple visual hallucinations, followed by hemiclonic seizures.7 Treatment with AED is necessary, and 50–60% of children become seizure free within 2–3 years.4
Childhood absence epilepsy accounts for 8–15% of all childhood epilepsies.8 Age of onset is typically between 4 and 10 years old. Absence seizures are very brief, usually lasting only seconds; however, they can occur up to hundreds of times a day. They are characterised by behavioural arrest and impaired consciousness. Approximately 60% of children will have associated automatisms including eye blinking or lip smacking. The classical electroencephalogram (EEG) for absence epilepsy is 3 Hz spike and wave complexes. Many of these children have associated behavioural and cognitive impairments, including an increased prevalence of attention deficit hyperactivity disorder (ADHD).8 In total, 70% of cases respond to ethosuximide.8 In 60–80% of patients, remission will be achieved within 2–5 years of onset. Fifteen percent of children with absence epilepsy will develop juvenile myoclonic epilepsy (JME).4
JME includes 5–10% of epilepsy syndromes. It usually peaks between the ages of 13 and 15.8 Presentation includes multiple different seizure types including myoclonic jerks, generalised tonic-clonic seizures, and absence seizures. Seizures typically occur in the mornings, and sleep deprivation is a common trigger. JME is a lifelong syndrome with most patients needing to remain on AED indefinitely.8
Epileptic encephalopathy with continuous spike and wave in sleep (CSWS) presents in children 2–4 years old as focal seizures and developmental regression.4,5 The EEG is classic for generalised spike and wave during slow wave sleep. The seizures and abnormal EEG typically resolve by age 9.4 The severity of cognitive decline and developmental regression is variable; however, it can affect behavioural, cognitive, language, and social modalities. Aetiology remains largely unknown, with genetic mutations accounting for approximately 17% of cases.4
Landau Kleffner syndrome (LKS) is an unusual childhood epilepsy syndrome which is described as an acquired epileptic aphasia. It typically presents in previously healthy children between the ages of 3 and 10 years old as significant, expressive, and receptive language regression. Children also develop seizures, which typically resolve by the age of 15.5 Recent research has supported a missense mutation in the GRIN2A gene, involved in NMDA receptor activation, as a possible aetiology for the syndrome.9
Lennox Gastaut syndrome (LGS) is one of the most devastating childhood epilepsy syndromes, and one of the most variable. LGS is characterised by multiple types of seizures, including drop attacks, cognitive and developmental regression, and generalised slow spike and wave complexes on EEG.10,11 Given the variable presentation of LGS, there are likely many underlying aetiologies. Infantile spasm, one of the most common epilepsy syndromes in the first year of life, is characterised by clusters of spasms, developmental regression, and hypsarrhythmia on the EEG; it may develop into LGS in approximately one third of cases.11
Dravet syndrome, also known as severe myoclonic epilepsy of infancy (SMEI), is an intractable epilepsy syndrome which usually presents in the first year of life.11 Infants will typically present with febrile seizures and then proceed to develop frequent afebrile seizures, developmental delays, and cognitive impairments.10,11 Approximately 80% of cases are caused by mutations in the SCN1A gene, which is involved in the voltage-gated sodium channel NaV1.1.10,11 Generalised epilepsy with febrile seizures plus (GEFS+) may also be caused by mutations in the SCN1A gene and is characterised by febrile seizures which continue into older childhood, and then progress into afebrile generalised seizures in adolescence.5
Ohtahara syndrome, also known as early infantile epileptic encephalopathy (EIEE), is one of the earliest epilepsy syndromes to present and can be diagnosed as early as the first week of life. The EEG is characterised by a burst suppression pattern. Seizures are typically intractable and do not respond to AED.10,11 EIEE has a very poor prognosis, associated with severe global developmental delay.10
OTHER AETIOLOGIES OF SEIZURE IN CHILDREN
While there are many known epilepsy syndromes, there are a multitude of factors which may lead to the development of seizures in children. Outside of epilepsy there is about a 1% lifetime risk of a single unprovoked seizure, and about a 6% lifetime risk of a provoked seizure.12 Typically, provoked seizures have little chance of recurring; however, febrile seizures may be an exception. Febrile seizures are the most common type of seizure, affecting 2–6% of the population.13 Three to four percent of children will have at least one febrile seizure and 20–40% of children will have a recurrence.12, 13 Febrile seizures are thought to be multifactorial, provoked by fever, genetic susceptibility, and the stage of brain maturation in young children.12 The risk of developing epilepsy after a simple febrile seizure is approximately 2% and up to 6% after a complex febrile seizure.13,14
Seizure following traumatic brain injury (TBI) is another example of provoked seizure and is a common complication after severe TBI in children. Post traumatic seizure (PTS) occurs in roughly 16% of 14–17-year-olds with accidental severe TBI and in up to 36% of children <2 years old.15 PTS increases up to 57% in the setting of subdural haemorrhage (SDH) and non-accidental trauma in younger children. Both SDH and non-accidental trauma have each been shown to increase the risk of PTS 2-fold.15 The risk of PTS decreases with age.15 A provoked seizure can be caused by any interruption to the functioning of the cerebral cortex, and the triggers are many. These include metabolic abnormalities such as electrolyte derangements, chemical or inflammatory irritation in the setting of meningitis or encephalitis, ischaemia, haemorrhage, or even concussion.16 Seizures can be caused by progressive neurological disorders or tumours. They can also result from perinatal asphyxia, hypoxic insult, or from abnormal neuronal proliferation and migration.16 Neuronal migration disorders such as heterotopia, lissencephaly, schizencephaly, and polymicrogyria have all been shown to cause seizures.17
The incidence and prevalence of epilepsy is higher in developing countries than in more developed countries.18,19 In sub-Saharan Africa and in rural India, it has been shown that the most common risk factors for seizure include family history of seizure, trauma or asphyxia at birth, head trauma, and central nervous system infection.18,19 In sub-Saharan Africa, parasitic infections, such as neurocysticercosis, cysticercosis, and toxocariasis are the most common infectious cause of seizures.1 Roughly 80–85% of epilepsy patients in the developing world are not treated effectively. This is largely because of limited financial resources, as well as cultural and social stigmas surrounding seizure disorders.18
It is widely accepted that the risk of recurring seizures after a first unprovoked seizure is between 32% and 69%.20 Recently, six studies were analysed, which included 815 neurologically and developmentally normal children, and the rate of seizure recurrence was found to be 45% within 3 years.20 Many causes of epilepsy have been identified; however, in at least 50% of children with epilepsy the cause remains unknown.12 It has been suggested that epilepsies can be divided into three categories based on aetiology: unknown, genetic, and structural/metabolic.21 In recent years the genetic basis of epilepsy has garnered much attention and research.
GENETIC BASIS OF EPILEPSY
New genes, mutations, and variants involved in epilepsy are being identified almost daily. Most of the genes identified encode for neurotransmitter receptors, ion channels, molecules involved in intracellular signalling, or proteins involved in synaptic structure.22 Up to 40% of childhood epilepsies are thought to have some component of genetic involvement.23 Mutations in genes, leading to dysfunction in sodium, chloride, and potassium channels in neurons, have been well established to have a role in many types of epilepsy.24 Multiple epilepsy syndromes have been identified which result from mutations causing gain or loss of function in ion channels (Table 1).23-29 As new candidate genes in epilepsy are identified, new technologies in genetic testing are becoming available and more accessible, making the molecular diagnosis of epilepsy increasingly relevant to researchers, physicians, patients, and their families. Next-generation sequencing has allowed for the rapid sequencing of large amounts of DNA, making it possible to test many genes at once, or even the entire genome.30 Comparative genomic hybridisation (CGH) is used to detect copy number variants (CNV), including deletions, duplications, and complex rearrangements.31 In patients with epilepsy and developmental delay, abnormalities are found with CGH in up to 23.5% of cases.30 It has been shown that CNV involving chromosomal regions 1q21.1, 15q11.2, 15q13.3, 16p13.11, 16p11.2, and 22q11.2 are associated with epilepsy, intellectual disability, and autism.32,33 Recently, a large cohort study looked at CNV data of 1,255 patients with pre-existing CGH or single nucleotide polymorphism array, who had epilepsy in addition to other comorbid conditions such as intellectual disability, autism, or other neurological features. Of the 1,097 patients included in the study, 12.7% were identified as having pathogenic or possible pathogenic mutations.34 CGH is relatively inexpensive and results are usually available within 2–4 weeks, increasing the value in clinical settings.
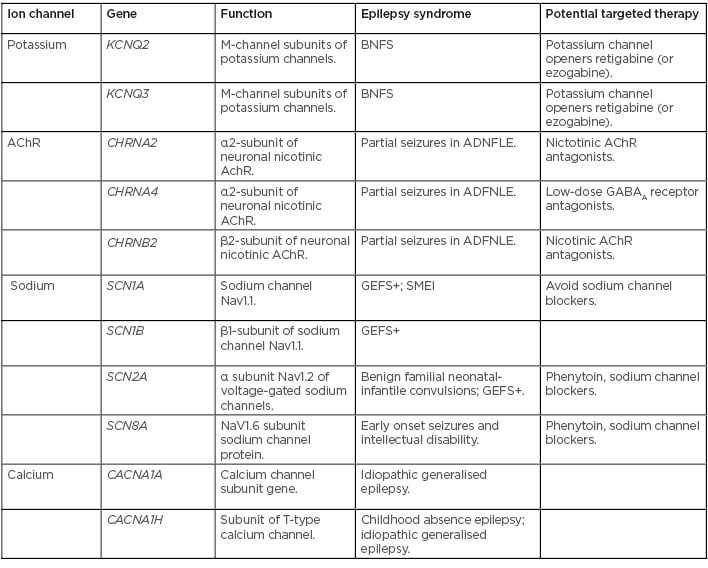
Table 1: Ion channel mutations identified in epilepsy syndromes.23-29
AChR: acetylcholine receptor; ADFNLE: partial seizures in autosomal dominant nocturnal frontal lobe epilepsy; BNFS: benign familial neonatal seizures; GABAA: γ-aminobutyric acid A; GEFS+: generalised epilepsy with febrile seizures plus; SMEI: severe myoclonic epilepsy of infancy.
TREATMENT
With increased availability of genetic testing and new mutations being identified, there is hope for new and more effective treatment of epilepsy in children. The standard of care and first-line treatment is AED therapy. There are currently >20 available AED, and choosing the appropriate medication largely depends on age and seizure type (Table 2).35-37 Approximately half of the patients become seizure-free on their first AED, and about two thirds will achieve seizure freedom with the use of AED.37 This leaves approximately 30% of patients with medication-refractory epilepsy. The ILAE defines drug-resistant epilepsy as “failure of adequate trials of two tolerated, appropriately chosen and used AED schedules (whether as monotherapies or in combination) to achieve sustained seizure freedom.”38 In addition to AED therapy there are other modalities available in the treatment of seizures.
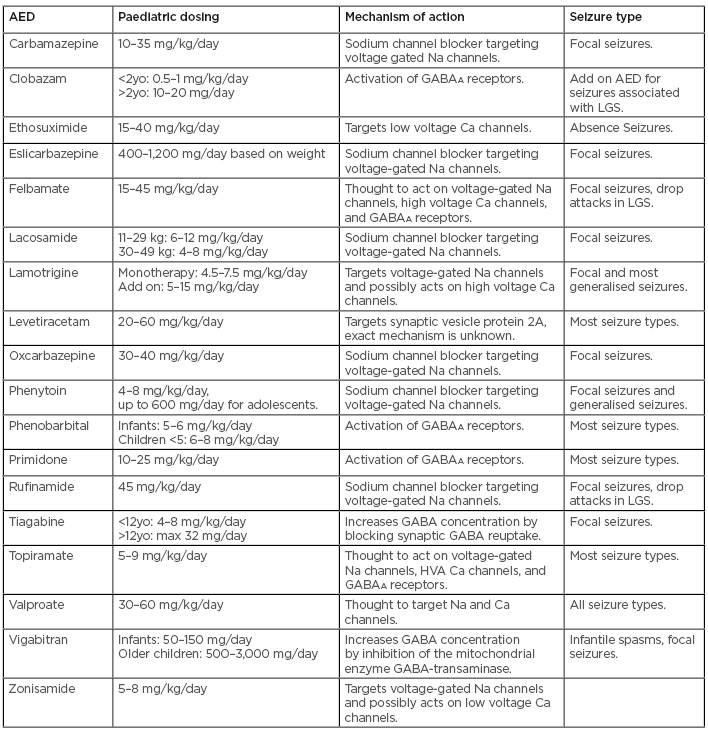
Table 2: Commonly used antiepileptic drugs in paediatric patients.35-37
AED: antiepileptic drug; GABAA: γ-aminobutyric acid A; HVA: high voltage activated; LGS: Lennox Gastaut syndrome; yo: years old.
The ketogenic diet has been proven to be an effective treatment for children with drug-resistant epilepsy. 37,39 The classical ketogenic diet consists of a diet composed of 90% fats, 7% proteins, and 3% carbohydrates.40 Today, there are multiple variations of the ketogenic diet consisting of varying ratios of fats to proteins and carbohydrates. A 2006 review of 15 studies on the ketogenic diet found a >50% reduction in seizure frequency in approximately one third of children with medication-refractory epilepsy on the ketogenic diet.39 For patients with glucose transporter type1 deficiency syndrome (Glut1DS), in which a mutation in the SLC2A gene causes impaired transport of glucose across the blood-brain barrier resulting in seizures; and in those with pyruvate dehydrogenase deficiency (PDHD), in which pyruvate cannot be metabolised into acetyl-CoA also resulting in seizures, the ketogenic diet is first-line therapy.37,40 In most patients with intractable epilepsy, the mechanism by which the ketogenic diet decreases seizure activity remains largely unknown.
Neuromodulation is another treatment modality available to children with medication-refractory epilepsy. In 1997 the U.S. Food and Drug Administration (FDA) approved the vagal nerve stimulator (VNS) as an adjuvant treatment for patients aged 12 years and older with refractory epilepsy.41 In 2017, the VNS was approved by the FDA for children age 4 years and older with medication-refractory focal onset seizures.41 A number of studies have shown that about half of paediatric patients achieve >50% reduction in seizure frequency.37,41 However, <5% of patients with a VNS became seizure-free.37 In addition to VNS, there are newer available neurostimulation therapies, which include deep brain stimulation of the anterior nucleus of the thalamus, responsive neurostimulation, trigeminal nerve stimulation, and transcutaneous vagus nerve stimulation.37,41,42
Another available treatment modality for intractable epilepsy is surgery. Over the past decade, there has been significant progress in the imaging and surgical techniques available. The most basic type of epilepsy surgery is the lesionectomy. Candidates for lesionectomy must have failed two or more AED and have epilepsy that is localised to a well-defined region based on EEG and radiological findings.43 Other available surgical treatments include lobectomy, multilobar resection, hemispherectomy, or disconnection of hemispheres.43 Recently there have been some trials and advancements in minimally invasive surgical techniques. MRI-guided laser interstitial thermal therapy, radiofrequency thermocoagulation, and magnetic resonance guided focussed ultrasound surgery are all relatively new techniques which allow for the ablation of a desired area of tissue.44-46 Further studies are needed to validate the efficacy and safety of these new non-invasive techniques.
In recent years, cannabidiol (CBD) has emerged as a new potential AED. CBD has been found to be most effective in refractory epilepsy syndromes such as Lennox Gastaut and Dravet syndromes.47 Epidiolex is the first CBD medication approved by the FDA for treatment of LGS and Dravet.41 Some of the suspected mechanisms by which CBD controls seizures include modulating neuronal excitability, antioxidant activity, and anti-inflammatory effects;47,48 however, the exact mechanism of action remains unknown. Although CBD may provide a promising alternative treatment for refractory epilepsy, recent studies have shown that up 55% of patients discontinue treatment and up to 94% report unwanted side effects, such as somnolence, decreased appetite, diarrhoea, and vomiting.49,50 The antiseizure activity of CBD has been proven; however, the long-term safety and efficacy of the medication is still under investigation.
TARGETED THERAPIES
The newest advancement in the treatment of paediatric epilepsies is based around the idea of precision medicine or targeted therapy. The continued identification of new genes and mutations underlying these syndromes is allowing for the development of individualised therapies for patients based on the molecular pathophysiology of the mutation found in a specific child. Targeted treatment can range from the use of ketogenic diet in Glut1DS, avoiding AED that act on sodium channels in patients with mutations in SCN1A, or the use of a specific drug that targets a specific mutation.
Pharmacogenomics uses the principle that an individual’s genetic background affects their response, including efficacy and adverse reactions, to a specific drug.28,37 There has been progress in identifying genomic predictors of response to various AED, which also has a role in individualising treatment for epilepsy patients. It has been established that cytochrome P450 enzymes are involved in the metabolism of many AED. In particular, it has been shown that polymorphisms in CYP2C9, CYP2C19, CYP2D6, and CYP3A4 can affect individual AED metabolism.28,51Patients with pathogenic CYP2C19 polymorphisms can have reduced clearance of phenobarbital, and the metabolism of valproic acid is altered with polymorphisms in CYP2C9.28,52 It has also been shown that the CYP3A4*1B variant in Mexican children with epilepsy resulted in multidrug resistance to AED.51 There is also evidence that a specific G to A polymorphism in the SCN1A gene correlates with needing higher doses of carbamazepine for therapeutic effect.28,53
The most recent advances in precision medicine are those which are identifying treatments for the damaged products resulting from specific gene mutations. Many of these therapies are still under research or in trial; however, there is much promise for the future of targeted medications. Everolimus is a medication which targets the mTOR signalling pathway, which regulates cell growth and differentiation in the brain.48 Tuberous sclerosis has been identified as resulting from mutations in the regulator proteins of mTOR. In both mouse models and clinical trials, everolimus was shown to be effective in treating seizures in tuberous sclerosis.48 As discussed above, mutations in GRIN2A, which encodes for a subunit of the NMDA receptor, have been identified as epilepsy syndromes such as LKS. There have been multiple open label trials investigating the effects of NMDA receptor antagonists as a possible seizure treatment.28,48 Memantine resulted in reduced seizure activity in a child with early-onset epilepsy with a GRIN2A missense mutation;28,54 however, in another child with a different missense mutation in GRIN2A memantine was not effective.28 A gain of function mutation in the potassium channel gene KCNT1 was noted to cause the early-onset epilepsy syndrome: epilepsy of infancy with migrating focal seizures. The anti-arrhythmic drug quinidine is a partial antagonist of KCNT1, and has been shown in laboratory studies of Xenopus oocytes, and in one confirmed case of a child with migrating focal epilepsy, to significantly reduce seizure activity.28,48,54 Mutations in KCNQ2 have been identified in a wide range of childhood epilepsies. Retigabine (or ezogabine) is a drug which acts as a positive modulator of KCNQ2 potassium channels, and it is the first neuronal potassium channel opener used in the treatment of epilepsy. 28,54 As new genes are identified the opportunity for new and individualised treatment grows. Recently, loss of function mutations in DEPDC5 (Disheveled, Egl-10, and Pleckstrin Domain Containing Protein 5) have been reported in a variety of genetic focal epilepsy syndromes.54 Research has also shown that mutations in PRICKLE proteins, which are involved in cell polarity signalling pathways, cause seizures in humans, mice, zebrafish, and flies.54 These newly identified genes may provide additional targets in the development of new precision therapies. The number of genes yet to be identified in paediatric epilepsy is unlimited. Large cohort studies, such as EpiPGX, are using genome-wide analysis in large populations of patients with epilepsy syndromes to identify genetic biomarkers with the goals of improving use of our current AED and identifying new targets for therapies.54,55
CONCLUSION
Paediatric epilepsy is one of the most common neurological disorders affecting children across the world. While almost two thirds of paediatric epilepsy patients are efficiently managed with available AED, the remaining one third rely on alternative therapies such ketogenic diet, neuromodulators, and surgery. The ongoing research in genetics and neurobiology holds promise for the development of individualised treatment with optimisation of our current AED, and the development of new targeted therapies. The possibilities remain endless; however, determining the safety and efficacy of new therapies may prove to be challenging and costly.






