
This symposium took place on 20th September 2021, as part of the World Muscle Society (WMS) 2021 Virtual Congress
Chairpeople: Laurent Servais,¹ Perry Shieh²
Speakers: Laurent Servais, Richard Finkel,³ Elena Mazzone,⁴ Perry Shieh, Crystal Proud⁵
1.MDUK Oxford Neuromuscular Centre, UK
2.Neurology and Paediatrics, University of California Los Angeles (UCLA), Los Angeles, USA
3.Center for Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, Tennessee, USA
4.Catholic University, Rome, Italy
5.Neurology department and Spinal Muscular Atrophy Center, Children’s Hospital of The King’s Daughters, Norfolk, Virginia, USA
Disclosure: Servais has received research funding from Biogen, AveXis, and Hoffmann-La Roche; and has received fees for symposia, lectures, and scientific advisory boards from Biogen, Hoffmann-La Roche, AveXis, Cytokinetics, and SMA Europe. Shieh has received fees for lecture and consultancy services from Alexion, AveXis, Biogen, CSL Behring, Genentech, Grifols, Roche, and Sarepta Therapeutics; and fees for research funding from AMO Pharma, Astellas, AveXis, Biogen, Catalyst, Fulcrum, Pfizer, PTC, ReveraGen, Sanofi, Sarepta, Santhera, and Solid. Finkel has received fees for research support from AveXis/Novartis, Biogen/Ionis, Capricor, Catabasis, ReveraGen, Roche/Genentech, Scholar Rock, NIH, Muscular Dystrophy Association, and the Charcot-Marie-Tooth Association; has received fees for symposia, lectures, and scientific advisory boards from AveXis/Novartis, Biogen/Ionis, Capricor, Catabasis, Cure SMA, Neurogene, ReveraGen, Roche/Genentech, Sarepta, SMA REACH UK, and Summit; and receives license fees for co-development of the Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disorders (CHOP INTEND). Mazzone has received consultancy and advisory boards fees from Biogen, Hoffmann-La Roche, Scholar Rock, Audentes Therapeutics, and AveXis and Novartis. Proud has received fees for consultancy, advisory service, or lectureships from AveXis/Novartis Gene Therapies, Biogen, Genentech/Roche, Sarepta Therapeutics, and Scholar Rock; and fees for research funding from AMO Pharma, Astellas, AveXis/Novartis Gene Therapies, Biogen, CSL Behring, FibroGen, Pfizer, PTC, Sarepta Therapeutics, and Scholar Rock.
Acknowledgements: Writing assistance was provided by Bronwyn Boyes, London, UK.
Support: The publication of this article was funded by F. Hoffmann-La Roche Ltd., while the symposium was funded by F. Hoffmann-La Roche Ltd. and co-funded by Sarepta Therapeutics. The views and opinions expressed are those of the authors and not necessarily the companies.
Citation: EMJ Neurol. 2022;10[Suppl 1]:2-15.
Meeting Summary
Both spinal muscular atrophy (SMA) and Duchenne muscular dystrophy (DMD) are monogenic neuromuscular diseases, which cause progressive proximal-to-distal muscular weakness, leading to loss of motor function and related pulmonary and musculoskeletal co-morbidities and reduced survival.1 Classic SMA is an autosomal recessive disorder affecting motor neurons, typically caused by homozygous deletions of the SMN1 on chromosome 5q, resulting in a deficiency of survival motor neuron protein, critical for motor neuron function and survival.2 DMD is an X-linked recessive muscle disease most often due to exon deletions, but also duplications and mutations in the DMD gene that encodes the dystrophin protein.3 Worldwide, SMA and DMD are the leading causes of neuromuscular disorders affecting children, which has led to active and innovative therapeutic research.4 There are multiple promising novel therapies available currently and with more on the horizon, which have immense potential to transform this field and prolong the functional independence and lifespan for individuals with SMA and DMD.1
Spinal Muscular Atrophy in the Spotlight: From New-Borns to Adults
The key takeaway messages from two previously held symposia at the 16th International Congress on Neuromuscular Diseases (ICNMD) and the 7th Congress of the European Academy of Neurology (EAN) in 2021 were: while motor function is an important outcome for infants with early infantile onset SMA (likely Type 1), improvements beyond motor function and survival are meaningful and can have a substantial impact on the daily lives of these infants and their caregivers.5 Similarly, for individuals with later-onset SMA (likely Type 2 or 3), improvements in motor function, however small, and disease stabilisation may maintain independence and can have a meaningful impact on activity of daily living.6 There is a knowledge gap to identify and evaluate the response to treatment in adults with late-onset SMA, owing to the progressive nature of SMA.6 To complete this symposia series, the World Muscle Society (WMS) symposium aimed at focusing on pre-symptomatic patients and remaining unmet needs in SMA care.
Shining a Light on Pre-symptomatic Spinal Muscular Atrophy
Richard Finkel and Elena Mazzone
Pre-symptomatic SMA can be defined as those individuals identified by new-born screening or are at risk from a positive family history, as opposed to having noticeable symptoms. These individuals may have normal development but may be affected by neuronal loss before birth and are at risk to develop symptoms later in life, especially in those with two copies of SMN2 (a paralogous gene that also encodes the survival of motor neuron [SMN] protein but producing lower levels of the functional protein). When symptoms develop, patients are diagnosed as SMA Type 1, 2, or 3, depending on age of onset and severity of symptoms.
Untreated Type 1 Spinal Muscle Atrophy
Most infants (approximately 60%) diagnosed with SMA have Type 1 SMA and, of those, the vast majority have two copies of SMN2.7 Untreated infants with Type 1 SMA with two copies of SMN2 do not achieve typical milestones compared to healthy infants (Figure 1). 8-14
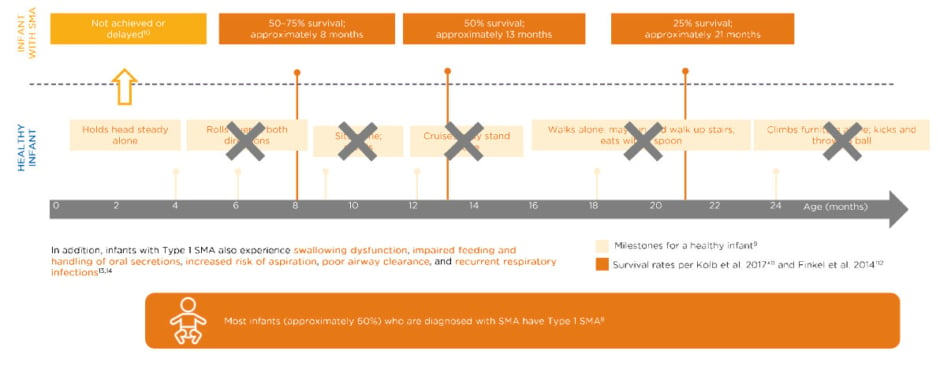
Figure 1: Untreated infants with Type 1 spinal muscular atrophy with two SMN2 copies never achieve key milestones.8-14
Data extrapolated from Figure 1B in Finkel RS et al.12
*No death or no intubation; n=20.
†No death or no need for ≥16 hour/day ventilation continuously for ≥2 weeks, in the absence of an acute reversible illness; n=34.
SMN: survival of motor neuron.
These infants may hold their head steady by 4 months, one of the Hammersmith Infant Neurological Examination Section 2 (HINE-2) milestones,9,10 but may never roll over, sit, cruise, walk, or climb furniture. The typical survival of such a patient at about 8 months of age is only 50–75% and that declines to about 50% at 13 months, and 25% survival in 21-month-olds.10
Outcome Measures
Since the introduction of disease-modifying treatments (DMT), primary outcome measures have expanded the focus beyond survival and ventilation to various motor function assessments.5,6 Several scales are used to assess motor disability and its progression in SMA. The choice of a scale should be based on the patient’s age, SMA type, and current motor function status (Table 1).
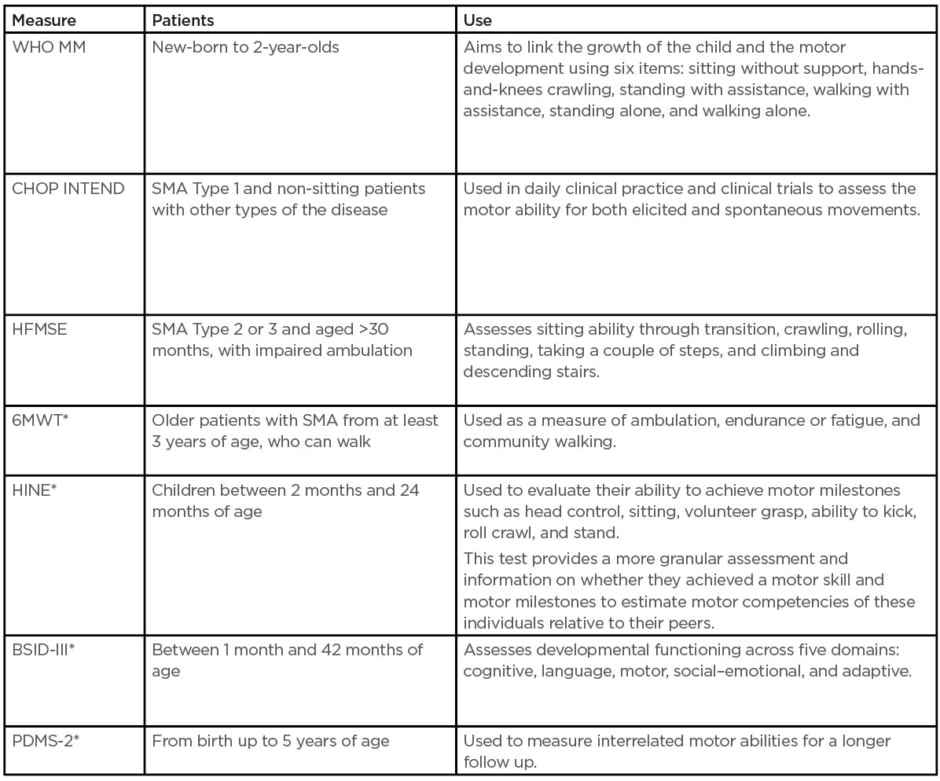
Table 1: Outcome measures used in clinical trials and practice.15
*Indicates there is normative data for typically developing children.
BSID-III: Bayley Scales of Infant and Toddler Development; CHOP INTEND: Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disorders; HFMSE: Hammersmith Functional Motor Scale Expanded for spinal muscular atrophy; HINE: Hammersmith Infant Neurologic Examination; PDMS-2: Peabody Developmental Motor Scale 2; SMA: spinal muscular atrophy; WHO MM: World Health Organization Motor Milestones; 6MWT: 6-minute walk test.
*Indicates there is normative data for typically developing children.
BSID-III: Bayley Scales of Infant and Toddler Development; CHOP INTEND: Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disorders; HFMSE: Hammersmith Functional Motor Scale Expanded for spinal muscular atrophy; HINE: Hammersmith Infant Neurologic Examination; PDMS-2: Peabody Developmental Motor Scale 2; SMA: spinal muscular atrophy; WHO MM: World Health Organization Motor Milestones; 6MWT: 6-minute walk test.
These assessments are useful in identifying any developmental delay and following patients in early intervention. They are also used to plan and monitor progress after initiation of any intervention programme. These instruments are based on linear hierarchical obtainment of motor skills, so they also refer to normative data. They can address gross and fine motor skill and are also able to capture other aspects in psychomotor development.15 Additional measures that should be considered in subpopulations and in certain aspects include bulbar function, respiratory function, fatigue, and compound muscle action potential or neurofilament.7,15,16
Panel Discussion
What should we do once a pre-symptomatic patient with spinal muscle atrophy is being treated?
Mazzone said that studies show that infants identified and treated early with DMTs tend to do very well, providing the highest magnitude of response and chance for normal development. Clinicians see many individuals who have psychomotor development, which does not differ too much from healthy peers. There is still the need to understand quantitative measurements, qualitative measurements, and to see the timeframe in which all of these motor milestones are achieved and if they are maintained over time. Data that goes beyond age-appropriate timeframes and gross motor function to look at fine motor function are needed, and for a longer follow-up.
What test could help ensure that younger patients are completely developing normally?
Mazzone said that is necessary to look at infant and toddler skills that cover development, looking at more granular achievement of motor skills and are age appropriate. The Bayley Scales of Infant Development (BSID-III) and Peabody Development Motor Scales 2 (PDMS-2) are used in trials. These scales can follow-up to 42 months or 5 years of age, respectively, and can capture a broader assessment of fine and gross motor skills but also have some additional value looking at cognition, language, expression, social, and behavioural assessments. One of the challenges has been what to assess, how to assess this, and how to address changes over time as patients mature. Measuring fatigue is also important, so sustainability and sustainability through growth at longer follow-ups.
CLINICAL TRIALS IN PRE-SYMPTOMATIC INFANTS
There are three key trials investigating treatment of pre-symptomatic infants (Table 2).17-25
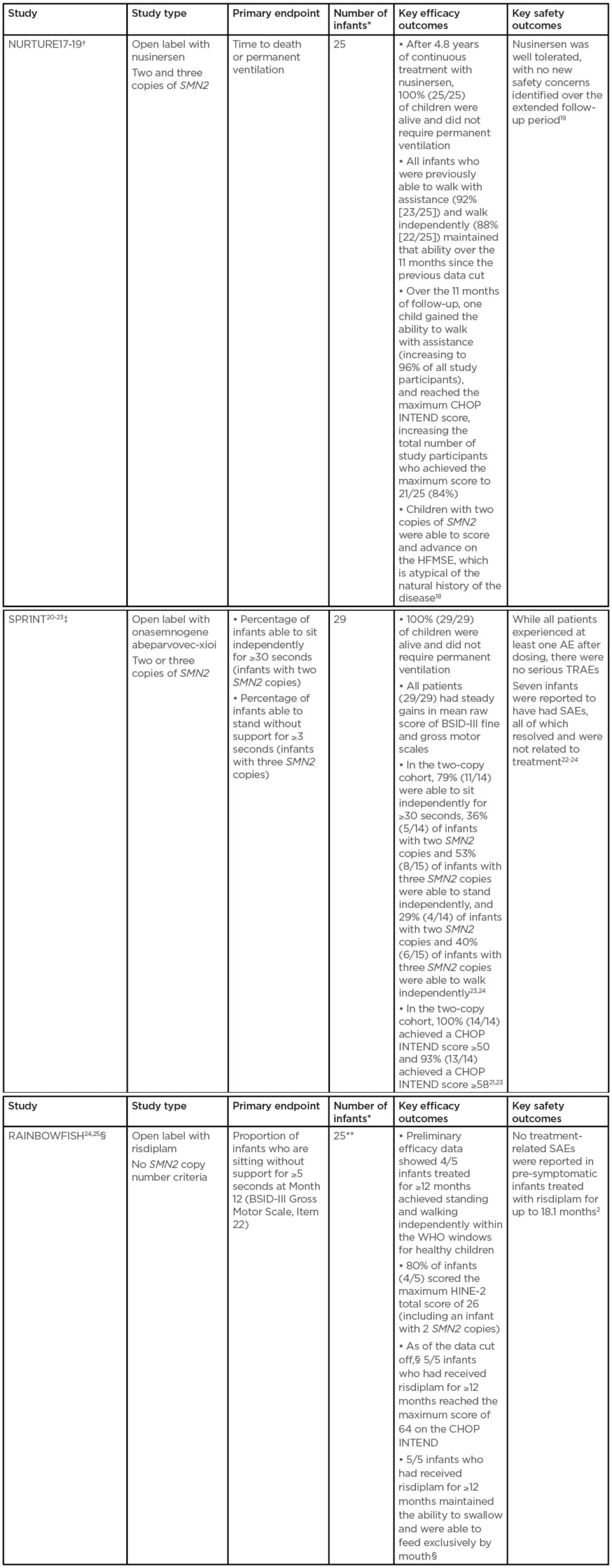
Table 2: Table 2: Key studies investigating treatment of pre-symptomatic infants.
All three studies limit inclusion to infants up to 42 days old at first dose.
*With genetically diagnosed and pre-symptomatic SMA.
†At data cut-off: February 2020.
‡At data cut-off: 11th June 2020.
§At data cut-off: 20th February 2020. Five infants have been treated for ≥12 months (preliminary efficacy data are available for these infants), includes two infants with two SMN2 copies and three infants with more than two SMN2 copies. Three infants have been treated for ≥6–<12 months. Four infants have been treated for <6 months.
**Preliminary data for five infants who have been treated for ≥12 months.
AE: adverse events; BSID-III: Bayley Scales of Infant and Toddler Development; CHOP INTEND: Children’s Hospital of Philadelphia Infant Test for Neuromuscular Disorders; SAE: serious adverse event; SMA: spinal muscular atrophy; TRAE: treatment-related adverse event; WHO: World Health Organization.
Carrying the Torch: How Do We Build on the Progress Made So Far?
The published clinical trial and emerging real-world data on patients with SMA treated with these DMTs presents compelling evidence of improved patient outcomes for those with two or three copies of SMN2, especially so for those treated in the pre-symptomatic state. However, significant knowledge gaps remain regarding the potential for response to these drugs in a broader population of patients with SMA. The panel discussed some of these key unmet needs.
Panel Discussion
Please comment on treatment of pre-symptomatic patients with one or four (rather than two or three) copies of SMN2.
Finkel stated that there is strong evidence from clinical trials to support the urgent treatment of babies with two or three copies of SMN2. However, patients with one or four copies have not generally been included in these trials (a key exception is the ongoing RAINBOWFISH trial with risdiplam),24,25 and data to inform how to address these patients are slowly emerging. Two recent case reports have been published regarding babies with a single copy of the SMN2 gene.26,27 In one case, the child made modest motor improvements in response to sequential treatment;26 however, this improvement plateaued, and the patient required a tracheostomy and feeding tube. She was discharged without regression of function but remains profoundly weak. Thus, whether treatment is clinically meaningful is debatable. At the other end of the spectrum, those with four copies, that data is still emerging as well. The obligation is on clinicians to very carefully monitor these patients clinically, and to gather biomarkers to develop a good argument as to when these babies should be treated. It is not a matter of if babies with four copies are expected to become symptomatic; the question is when and with what urgency.
What expectations would you discuss with a parent of an infant with three copies of SMN2 identified through new-born screening?
Finkel said that for babies with three copies, the emerging data from all three studies outlined in Table 1 is really remarkable and these babies are uniformly doing well, generally following normal trajectories in motor function, and also in their bulbar function, feeding, communication, and respiratory function. What is currently unknown is the durability of these effects during growth and development with increased demands on their muscle mass. Therefore, clinicians have to be cautious when talking to parents. Babies with two copies are also doing remarkably well, but in some cases lagging behind in the acquisition of motor milestones and a few needing feeding and/or respiratory support. Clinicians do not know if these babies will catch up over time or develop the need for additional levels of supportive care.
What are the current challenges to implementing new-born screening programmes, recognising that there are differences from country to country and sometimes even within a country?
Servais noted that the main challenge is actually to have a plan about what you are going to do with your patients with one, two, three, and four or more copies. This is very country-dependent and payer-dependent. The second thing is that you need multistakeholder involvement. If you want to start a pilot, you need to have on board the neuromuscular doctors, those involved in new-born screening, the nurses, the public, and the committee in charge of new-born screening involved if you want a pilot to be successful. Lastly, you need to have a plan in terms of timeline to be able to deliver the evidence that you need. In some countries, you just need to show that it is feasible and does not disrupt the overall new-born screening programme. In other countries, you will need negative predictive value of a negative test, and the positive predictive value of a positive test, which then needs a proper power calculation. Some countries will require health economic data.
It would be difficult to identify a baby but not have a treatment available for that baby. What are your thoughts?
Servais said that many of his patients have had a long diagnostic journey. Visiting a general practitioner or paediatrician first and enduring for years to try to find the diagnosis. The MRI or other exams that will be sometimes prescribed will be normal. They may have a wrong diagnosis, and sometimes they may be labelled as psychiatric. Finally, they get a diagnosis after a long diagnostic journey, during which they have lost quite a significant number of motor neurons. It would be a significant benefit to reduce the burden and the cost of this journey and to accelerate treatment access for these patients, with an earlier diagnosis. So, the rationale of not being able to treat the patients with four copies should not preclude the identification of these patients. Clinicians need to identify these patients and have a clear plan for the follow up of these patients and then treat them.
What is your vision of the future?
Finkel said that he cannot look 20 years into the future but perhaps it is feasible in the more immediate future, for the next few years. There are three wonderful drugs, but have all three really been optimised? There are ongoing studies looking at higher doses, intrathecal delivery, and emerging real-world evidence in a broader population of patients, both symptomatic and pre-symptomatic. There are also studies investigating sequential treatment or combinatorial treatment.
Does the burden of treatment always surpass the benefit in adult patients and how are these patients assessed?
Finkel stated that in the USA, about 40% of the prevalent population is untreated. So, there are three drugs, but almost half of this population (who are mostly adolescent or adult patients) are not electing to start a treatment. Clinicians need to understand their reasons. Is it due to limitations in health insurance coverage for patients with four copies of Type 3 SMA, reimbursement, related to the burden of repeated lumbar punctures, or do these individuals not understand the progressive nature of the disease? Clinicians need to educate this community and hopefully persuade them that there are good drugs, and they need to consider treatment to rescue as many of these motor neurons as possible and sustain their current level and maybe even improve it.
Conclusion
Laurent Servais
Servais concluded that DMTs, in combination with standard of care, have demonstrated a dramatic efficacy in changing the outcome in individuals with pre-symptomatic SMA (compared with natural history). Therefore, new-born screening programmes should be implemented globally so that individuals with SMA can be identified and treated as early as possible. There are remaining unmet needs for all people living with SMA, from new-borns to adults. These include the need to capture additional outcomes that are meaningful to individuals with SMA, and the urgency to identify and treat adults with late-onset SMA, who have not yet initiated treatment with DMTs. The SMA community must continue to work together to meet these remaining needs and further improve the lives of those living with SMA and of their caregivers.
CONNECTING THE DOTS BETWEEN NATURAL HISTORY AND CLINICAL ADVANCES IN DUCHENNE MUSCULAR DYSTROPHY
The Underlying Complexities of Duchenne Muscular Dystrophy: Lessons from Natural History
Crystal Proud
DMD is a rare, X-linked, fatal, neuromuscular disease that affects approximately one male in 3,500–5,000 births worldwide.28-30 It is characterised by intrinsic muscle inflammation, degradation, and fibrosis, which leads to progressive motor dysfunction.28,29,31-42 The life expectancy of individuals with DMD is significantly reduced compared to healthy individuals.26 DMD leads to a variable but progressive sequential pattern of muscle weakness that eventually causes loss of important functional milestones such as the ability to walk.28,29,31-42
Muntoni et al.43 evaluated 395 individuals with DMD, characterising their age, and corresponding North Star Ambulatory Assessment (NSAA), to demonstrate the heterogeneity of this group. Patients may see improvement around age 4 years, until a peak around age 6 years, with subsequent decline at a rate of about 3 units per year. This variability may be influenced by the intrinsic, as well as extrinsic factors. Intrinsic factors include genetic modifiers, such as polymorphisms, or the location of specific mutations and impacts on endogenous exon skipping.44 Extrinsic factors include access to care, resources, nutrition, and other supportive interventions, which may influence outcome.44 Muntoni et al. further highlighted this variability within the DMD population in the categorisation of trajectories into four classes. Faster progression was noted in patients in Classes 1 and 2, while a slower progression was seen in patients in Classes 3 and 4. Patients in Class 4 maintained an NSAA score of greater than 5, until at least 15 years of age. This variability is critical to consider when evaluating outcomes of treatment modalities.43
Early diagnosis is critical to initiate care for patients with DMD. Irreversible muscle damage starts during infancy and continues throughout the individual’s life in a progressive fashion.28,45 Genetic testing is considered standard of care to confirm a diagnosis of DMD.28 Given the progressive nature of DMD, treatment goals will be influenced by where the patient is along their journey with muscular dystrophy. If they are ambulatory, a treatment goal may be to delay the loss of ambulation, or to maintain the ability to stand. If they have lost ambulation, a treatment goal may be to preserve arm or hand function.28,46 Delaying or preventing disease progression remains a key unmet need of individuals with DMD.47 Management of DMD involves administration of corticosteroids.28 These have demonstrated the ability to prolong ambulation and have positive effects on scoliosis, cardiac and pulmonary function, as well as mortality.48-53 However, steroids are associated with adverse events. These include impacts on bone health, delayed puberty, weight gain, immune suppression, and behavioural changes.53,54
Some of the management of complications of DMD include medications, such as angiotensin-converting enzyme inhibitors and other heart failure treatments to optimise cardiac function,28 respiratory insufficiency is addressed through non-invasive or invasive ventilatory support, and airway clearance is facilitated through cough assist.28,55 Physical therapy works to address a range of motion, reduce fatigue, and accommodate to gross motor changes. Occupational therapy addresses the activities of daily living that are impacted by the patient’s muscular weakness and bracing may be utilised in attempts to prevent progressive contracture. If scoliosis is present, surgical intervention may be required.28,42,46 In addition to corticosteroid treatment, targeted therapies are available in certain countries for patients with specific mutations. Antisense therapies target certain skippable mutations to restore the reading frame and lead to a partially functional protein. These include eteplirsen for exon 51,28,56,57 golodirsen58 and viltolarsen59,60 for exon 53, and casimersen61 for exon 45 skip amenable mutations, which are currently available in the USA. Ataluren is an approved small molecule therapy that is approved in some countries for patients with nonsense mutations, leading to a stop codon.62,63 These therapies are mutation-specific and not applicable to all patients with DMD. Approximately 39–44% of individuals with DMD are treatable with these therapies.64
Proud presented two case studies to illustrate that age and functional status of the patient impact the decision making and treatment expectation.
Gene Therapy Clinical Trials for Duchenne Muscular Dystrophy and Considerations for Clinical Trial Design
Perry Shieh
There are several ongoing gene therapy studies for DMD that aim to deliver a shortened but potentially functional version of dystrophin.65-73 Shieh then focused on the delandistrogene moxeparvovec (SRP-9001; Sarepta Therapeutics, Cambridge, Massachusetts, USA) gene therapy clinical trial programme. The dystrophin gene is too large to be packaged into an adeno-associated virus (AAV) vector. The delandistrogene moxeparvovec construct is designed to provide micro-dystrophin expression in the skeletal and cardiac muscle and to be packaged in an AAV vector.64,74-77
There have been three studies that have enrolled patients treated with delandistrogene moxeparvovec. Study 101 was the original study and enrolled four boys with DMD.65,66 Study 102 is a two-part, randomised, double blind, placebo-controlled study.67 ENDEAVOR (Study 103) is an open-label, systemic gene delivery study to evaluate the safety and expression of delandistrogene moxeparvovec in 20 participants with DMD using commercial processes.78
The disease trajectories may have an impact on the outcomes of the NSAA. Enrolment of younger boys should assess the potential of early intervention to preserve muscle. Enrolment of older boys should assess the potential of preserving remaining function.43 It is important to have cohort stratification, where comparisons should be considered in participants who are anticipated to be in similar ‘phases’ of their muscular dystrophy journey (cohorts of similar age or functional status). It is also important to consider the trial duration. In younger participants, the observation of effect during motor function decline (around 7 years of age and older) may provide more distinction between treatment versus placebo groups. In older participants, observation of effect may require a duration that distinguishes treatment effect versus incremental declining function as described by natural history.Consideration of effect of intervention must distinguish between improvement versus stabilisation versus slowing the rate of decline. Stabilisation being the more realistic goal of gene therapy in DMD. If the investigational product is to promote stabilisation for example, divergence of functional abilities attributed to treatment may not occur until a natural decline in the placebo group.43
LOOKING AHEAD: CHALLENGES AND OPPORTUNITIES IN THE DELANDISTROGENE MOXEPARVOVEC MICRO-DYSTROPHIN CLINICAL DEVELOPMENT PROGRAMME
Conclusion
DMD is a progressive neuromuscular disorder with a clinically variable rate of progression. Preventing further disease progression represents the key unmet need for DMD; therefore, the development of novel therapeutic interventions and clinical trial endpoints are needed in this therapy area. Gene transfer therapy is being explored as a strategy to treat DMD. Future trials will utilise learnings from previous trials to advance the delandistrogene moxeparvovec clinical development programme.
Symposium Conclusion
Selecting the right endpoint for neuromuscular disease clinical trials is not inconsequential and is complicated by the heterogeneity of disease manifestation in DMD and SMA. This necessitates multiple endpoint specification to enable the capture of different ages, severity, and stages. The design of clinical trials, therefore, requires careful consideration. Another challenge is finding measures of function that cover the spectrum of disease severity and symptoms to ensure an appropriate measure of treatment benefit.






