Abstract
Acute kidney injury is a frequent complication of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), principally because of hypotension and decreased kidney perfusion secondary to haemodynamic or haemostatic factors, drug-induced nephrotoxicity, and cytokine storm syndrome related to sepsis. Additionally, several factors support the existence of SARS-CoV-2-associated nephropathy, such as early, new-onset proteinuria and haematuria in many patients, the identification of SARS-CoV-2 viral load in precisely defined kidney compartments, ultrastructural and immunohistochemical evidence of direct viral infection of the kidneys, and, most importantly, morphological alterations associated with cytopathic action induced by the virus. In addition, collapsing glomerulopathy that has been reported in patients of African American descent with underlying apolipoprotein L1 (APOL1) kidney risk alleles and SARS-CoV-2 infection is evidence of a distinct form of SARS-CoV-2-associated nephropathy, the APOL1-SARS-CoV2-associated nephropathy.
INTRODUCTION
A cluster of pneumonia cases associated with an increased risk of death, subsequently ascribed to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), was reported in the city of Wuhan, China, in December 2019.1 On 11th March 2020, the World Health Organization (WHO) declared SARS-CoV-2 a pandemic.2 In 80% of cases, the infected subjects are asymptomatic, or paucisymptomatic with mild symptoms characterised by fever, sore throat, dry cough, conjunctivitis, tiredness, diarrhoea, anosmia/hyposmia, and, in some cases, ageusia.3 15–20% of patients, approximately 1 week after the first appearance of symptoms, progress toward a more serious disease characterised initially by worsening dyspnoea, a clinical manifestation of interstitial pneumonia, with the need for mechanical ventilation.4,5
The involvement of the respiratory tract is not the only serious systemic complication that occurs during SARS-CoV-2 infection. Many organs and systems may be involved, resulting in a multiorgan dysfunction syndrome that is largely caused by the cytokine release syndrome.5,6 However, autopsies have shown that SARS-CoV-2 has an organotropism beyond the respiratory tract that includes the kidneys, heart, liver, and brain (Figure 1).7
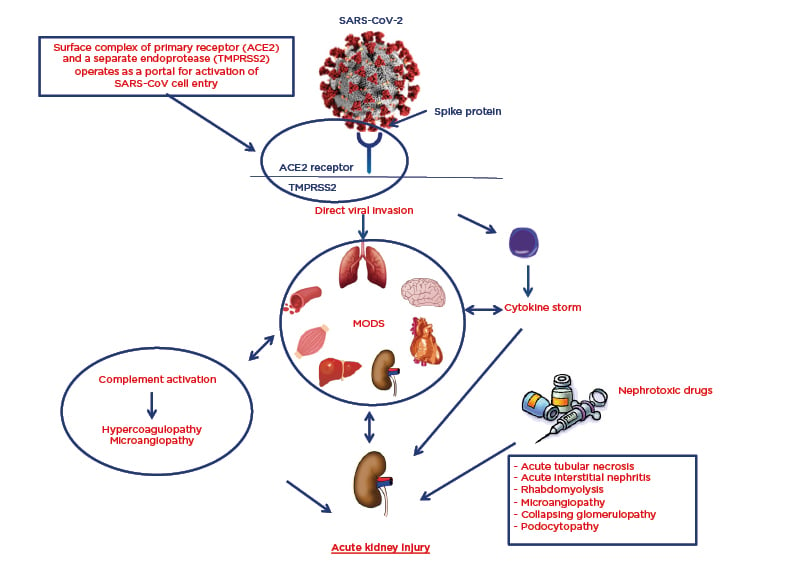
Figure 1: Pathophysiology of SARS-CoV-2 organotropism and mechanisms of acute kidney injury.
ACE2: angiotensin-converting enzyme 2; MODS: multiorgan dysfunction syndrome; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; TMPRSS2: transmembrane protease serine 2.
In this review, the authors report the epidemiological, clinical, pathophysiological, and histopathological data that support direct renal damage by SARS-CoV-2 that allows for the definition ‘SARS-CoV-2-nephropathy’.
EPIDEMIOLOGY AND CLINICAL MANIFESTATIONS OF RENAL INVOLVEMENT
Initial data suggested a low incidence (3–9%) of acute kidney injury (AKI) in patients with SARS-CoV-2.4,8-10 However, more recent data have reported the incidence of AKI in 27–37% of hospitalised patients.11,12 In a large population of patients hospitalised with SARS-CoV-2 in New York City, New York, USA (N=5,449), AKI developed in 37% of patients.12 According to the Kidney Disease Improving Global Outcomes (KDIGO) diagnostic criteria for AKI,13 47% of patients developed AKI Stage I, 22% AKI Stage II, and 31% AKI Stage III.12
15% of patients with AKI required haemodialysis treatment, and 97% of these were already on mechanical ventilation. AKI developed in 90% of patients on mechanical ventilation, while in noncritical patients it reduced to 22%.12 In addition, the temporal association between the beginning of mechanical ventilation and the development of AKI was very short (52% after 24 hours post-intubation).12
The development of AKI in patients with SARS-CoV-2 is an independent risk factor for in-hospital mortality, as demonstrated by two Chinese case studies.10,11 In the New York case series, the mortality of AKI patients was 35%, 26% of patients were discharged, and 39% of AKI patients were still hospitalised at the time of the study’s publication.12 The multivariate analysis of the New York case series highlighted the following factors as independent predictors of AKI development: advanced age, African American background, diabetes, hypertension, cardiovascular diseases, mechanical ventilation, and use of vasopressor drugs.12 In this analysis, the inhibitors of the renin–angiotensin–aldosterone system were not predictive of AKI,12 confirming the advice of many scientific societies and individual experts that their use should not be withdrawn for prophylactic reasons.14
Recently, Stewart et al.15 studied a cohort of 52 hospitalised children (aged 0–16 years) with confirmed SARS-CoV-2 infection and showed that 29% of these patients met the British Association of Paediatric Nephrology (BAPN) diagnostic criteria for AKI. Most cases of AKI occurred in those admitted to the paediatric intensive care unit (93%), and these patients were more likely to have presented with diarrhoea and vomiting, thereby suggesting prerenal involvement.
The exact mechanism of renal involvement and impairment in patients with SARS-CoV-2 is unclear. Postulated mechanisms include prerenal AKI during multiorgan dysfunction syndrome, especially in patients hospitalised in an intensive care unit and with previous reduced renal functional reserve (defined as a state of a normal or increased baseline serum creatinine and a reduced capacity of the kidney to increase glomerular filtration rate in response to certain physiological or pathological stimuli or demands because of the lack of compensation by residual nephrons); iatrogenic AKI by nephrotoxic drugs; release of proinflammatory cytokines, which could lead to acute interstitial nephritis; and direct virus cellular damage at the tubule cell and podocyte cell level (Figure 1).16,17
In favour of the hypothesis of a direct viral damage, and therefore of SARS-CoV-2 nephropathy, there are various clinical elements in addition to the histopathological ones. In one study, proteinuria and microhaematuria were noted in 44% and 27%, respectively, of 701 patients at the time of hospital admission.10 The same study reported a low incidence of intrahospital AKI (5%) in these patients, thus suggesting that, in the absence of histological data, the high percentage of urinary abnormalities found at admission were determined by glomerular damage related to the virus.10 Hong et al.,18 in an attempt to identify the incidence of early renal damage in 12 patients with SARS-CoV-2, reported albuminuria, without significant alterations in plasma creatinine, in five of the 12 patients.
However, the most significant data on glomerular damage has been linked to nonselective proteinuria, characterised by the presence of IgG and transferrin in the urine. Additionally, the finding of α1-microglobulin in the urine has been shown to indicate tubular damage.18 In contrast, the high percentage of microhaematuria (46%) and proteinuria (42%) found in patients with AKI in the New York series cannot undoubtedly indicate possible glomerular damage as the authors were unable to provide data on whether the patients had a bladder catheter at the time of urine collection.12
Furthermore, besides the urinary abnormalities found in many patients in the early stages of the disease, the detection of viral particles19 and viral RNA20 in the urine of some patients are indirect indicators of SARS-CoV-2 nephropathy.
PATHOPHYSIOLOGY OF THE RENAL ORGANOTROPISM
SARS-CoV-2, as well as SARS-CoV, invades cells through the binding of its spike protein with the angiotensin-converting enzyme 2 (ACE2) cell receptor.21 The ACE2 receptor is not only expressed on the cell membrane of Type 2 pneumocytes, but is also ubiquitous in the oral and nasal mucosa, nasopharynx, lymph nodes, thymus, bone marrow, spleen, liver, kidneys, brain, vessels, heart, and the entire gastrointestinal tract.22,23 In particular, the expression of ACE2 in the kidneys is higher than in the lungs.24,25 ACE2 is expressed mainly in the apical side of the proximal tubular cells and, to a minor degree, on the surface of the podocytes.25
It is hypothesised that the higher propensity of SARS-CoV-2 to induce serious infection at the bronchiole–alveolar level compared to other organs and systems is related to the fact that cellular infectivity does not depend only on the cellular expression of ACE2. The entry of SARS-CoV-2 into the cell through the ACE2 receptor is ‘helped’ by several proteases located on the cell surface, which are thought to have greater expression at the alveolar level. Meanwhile, other proteases facilitate the spread of the virus once it has penetrated the cell. In particular, transmembrane protease serine 2 (TMPRSS2), a member of the subfamily hepsin-TMPRSS, is a transmembrane proteolytic enzyme, which, although stoichiometrically separated, is structurally and functionally part of the ACE2 receptor (Figure 1). TMPRSS2 binds to the S1 subunit of the viral spike protein and detaches it from the S2 subunit. This enzymatic activity enhances the entrance of the virus into the cell through the ACE2 receptor by almost 100 times. This is because, after detachment, the viral S2 unit fuses with the cell membrane and forms a unit that transfers the viral content into the cytoplasm.26-28
In the kidneys, TMPRSS2 is more highly expressed in the distal tubular cells than in the proximal tubular cells.29-31 However, it remains to be determined whether other TMPRSS, such as TMPRSS4, 5, and 9, located at the level of the proximal tubular cells, can mediate the cellular entry of SARS-CoV-2.
The endothelial glomerular cells, which represent the first line of defence, can be infected directly by SARS-CoV-2, as has been demonstrated in a kidney transplant recipient with the help of electron microscopy.32 Subsequently, the virus could invade the podocytes; viral particles have been identified in the podocyte cytoplasm,33,34 and have been shown to pass through the Bowman’s space and infect the proximal and distal tubular cells, as demonstrated by the presence of viral particles also in the cytoplasm of proximal tubular cells.34,35
The difference in incidence of AKI observed between patients from Asia and patients from Western countries could be explained by a higher expression of ACE2 and proteases of the TMPRSS family at the renal level, as has been demonstrated in a recent transcriptomic analysis of 15 healthy human kidney samples.36
RENAL HISTOPATHOLOGY AND SARS-COV-2 NEPHROPATHY
The first renal histopathologic data came from 26 autopsies of SARS-CoV-2 infected patients.34 Under light microscopy, all the tissue samples analysed showed proximal acute tubular necrosis (ATN) of varying degrees, characterised by brush border loss and vacuolar degeneration. Occasional haemosiderin granules in tubular epithelium cells were observed in four patients with microhaematuria and clinical signs of rhabdomyolysis. The interstitium was free from cell infiltration. The glomeruli exhibited various alterations, characterised by signs of diabetic nephropathy in two patients with diabetes, and the presence of ischaemic glomeruli in 11 patients with hypertension. The glomerular and peritubular capillaries were obstructed by erythrocyte aggregates in the absence of platelets or fibrin thrombi in the lumen. At electron microscopy, coronavirus-like particles were present in the cytoplasm of proximal tubular cells and podocytes, but they were less evident at the level of distal tubular cells. Furthermore, with electron microscopy, it was possible to observe foot process effacement and occasional vacuolisation and detachment of the podocytes from the glomerular basement membrane, where cytoplasmic coronavirus-like particles were identified. Viral particles at the tubular level were identified with indirect immunofluorescence, which used antibodies directed against nucleoproteins common among betacoronaviruses. Meanwhile, the expression of ACE2 was shown to be upregulated, especially in sites with severe ATN, using immunohistochemistry.34 Taken together, these observations demonstrate that, in part, the pattern of ATN and the presence of new-onset proteinuria can be caused by direct viral damage.
In the study by Farkash et al.,35 the authors observed focal tubular isometric vacuolisation upon light microscopic examination, and upon ultrastructural analysis they revealed abundant viral forms within the tubular epithelial, which correlated directly to the areas of isometric vacuolisation; this could be a helpful diagnostic clue for the presence of direct renal infection.
Diao et al.,37 by analysing six samples of renal tissue post-mortem, found signs of ATN of varying degrees in all samples. In contrast, observations by Su et al.34 were that cell infiltrates were present at the tubulointerstitial level in five of the six samples analysed. Immunohistochemistry conducted by Diao et al.37 revealed that the vast majority of these cells were cluster of differentiation (CD)68+ macrophages, though CD8+ lymphocytes were also present to a moderate degree. Counter to this, the appearance of CD4+ lymphocytes and CD56+ natural killer cells were scarce. The direct SARS-CoV-2 cytopathic effect was detected in three samples by the presence of viral infection-associated syncytia. Under light microscopy, the glomeruli did not show significant alterations, except for some signs of focal segmental glomerulosclerosis with negative immunofluorescence, thought to be caused by immunoglobulin deposits, in three patients with hypertension.
Upon immunohistochemistry examination, expression of viral nucleocapsid antigens (limited to the cytoplasm of the tubular cells) was observed in all samples analysed. Several tubular cells that were positive for the expression of viral nucleocapsid antigens had also lost their contact with the tubular epithelium. Additionally, with immunohistochemistry, it was possible to highlight the presence of another important ‘actor’ that is co-responsible for kidney damage: the activation of the complement cascade was detected by a strong positivity for the membrane-attack complex (MAC; C5b-9) on tubules in all samples. A mild positivity of C5b-9 was found on glomeruli and capillaries in only two cases. A limitation of this paper was the absence of an ultrastructural investigation.37
The importance of the complement cascade in the pathogenesis of SARS-CoV-2 has been emphasised in a review by Noris et al.,38 and Jhaveri et al.39 were the first to describe a case of complement-mediated haemolytic uraemic syndrome with a widespread renal cortical necrosis.
In addition to systemic inflammatory damage affecting the vessels, as determined by the release of cytokines and the activation of the complement cascade, endothelial cells represent a preferential target of SARS-CoV-2. Varga et al.32 have demonstrated, with an ultrastructural examination, the presence of viral inclusions in glomerular endothelial cells.
To date, case reports of patients with African background with AKI, nephrotic proteinuria, and a histopathological picture of collapsing glomerulopathy have been published.33,40-46 The majority of the patients had an apolipoprotein L1 (APOL1) high-risk genotype,40-43 which also cannot be excluded for the patients who did not perform a genetic investigation.33,43-45 Patients with the G1 and G2 alleles of the APOL1 gene, in their vast majority of Sub-Saharan African descent, are known to be susceptible to chronic kidney disease and glomerulopathies such as focal segmental glomerulosclerosis and HIV-associated nephropathy. In recent years, the term APOL1-associated nephropathy has been widely used.47 The description of cases of collapsing glomerulopathy in patients with SARS-CoV-2 of African ethnicity and the link to APOL1 high-risk alleles could, therefore, explain why African descent represents an independent predictive factor for the development of AKI. A ‘two-hit’ combination of genetic predisposition and cytokine-mediated host response to SARS-CoV-2 infection could be the possible pathogenetic mechanism of collapsing glomerulopathy in these patients. However, Kissling et al.33 and Kadosh et al.45 have both independently identified viral inclusion bodies within the podocyte cytoplasm and the tubular epithelial cell cytoplasm, suggesting in these cases that the second hit could be caused directly by SARS-CoV-2.
Lazareth et al.46 have reported a case of collapsing glomerulopathy in a kidney transplant recipient of Sub-Saharan African origin, as well as SARS-CoV-2 infection with a low-risk donor of APOL1 genotype G0/G2, where the genotype of the recipient was G0/G0. This case report illustrates that a high-risk APOL1 donor background is not a prerequisite for severe podocyte injury to occur in kidney allografts in the context of SARS-CoV-2.
Sharma et al.48 reported more recent data from 10 patients with severe AKI (eight patients required haemodialysis). All biopsy samples showed varying degrees of ATN, and one patient had associated widespread myoglobin casts. In addition, two patients were found to have thrombotic microangiopathy, one had pauci-immune crescentic glomerulonephritis.48 In contrast with the above reports, immunohistochemical staining of kidney biopsy samples and ultrastructural examination were negative for direct renal infection.48
Similarly, Kudose et al.43 did not detect direct renal infection in 17 patients who had required kidney biopsy; 14 patients underwent native kidney biopsies, and three had allograft specimens. Among the 14 patients with a native kidney biopsy, five were diagnosed with collapsing glomerulopathy, one with minimal change disease, two with membranous glomerulopathy, one with crescentic transformation of lupus nephritis, one with anti-glomerular basement membrane nephritis, and four with isolated ATN.43
The most convincing demonstration of the renal tropism of SARS-CoV-2 comes from Puelles et al.,7 who first directly quantified the viral load in tissue samples of various organs from deceased patients with the use of in situ hybridisation. The highest viral loads were from respiratory tract samples, while lower values were found in the kidney, liver, heart, and brain.7 In particular, three of the six renal tissue samples analysed showed the presence of viral load in all anatomical compartments, but the preferred target of the virus was for glomerular cells.
CONCLUSION
Kidney damage during SARS-CoV-2 infection is multifactorial. The precarious haemodynamic state and the advanced age of many patients with AKI, together with the systemic inflammatory state, as an organic response to viral infection, represent the main causes of renal involvement. On the other hand, however, the preferential organotropism that the virus shows against the kidney should be underlined. The finding of viral RNA in the urine, and especially in samples of renal tissue from deceased patients with the use of in situ hybridisation, is almost certain proof of renal tropism, but does not confirm a direct cytopathic effect of SARS-CoV-2. The urinary abnormalities (proteinuria and microhaematuria) detected in the early stages of the disease, the identification of viral particles in the cytoplasm of proximal tubular cells, glomerular endothelial cells, and podocytes, and the positive immunohistochemical and immunofluorescence staining for viral antigens, represents indirect signs of renal cytopathic damage. Direct signs of cytopathic damage, and therefore of SARS-CoV-2 nephropathy, are represented by the morphological alterations found in renal cells with viral inclusion as focal tubular isometric vacuolisation; the presence of viral infection associated-syncytia; foot process effacement of podocytes; and the presence of tubular cells and podocytes detached from their supporting membranes.
The presence of a histopathological pattern of collapsing glomerulopathy in patients of Sub-Saharan African ethnicity carrying the APOL1 high-risk alleles also allows for the use the term APOL1-SARS-CoV-2-associated nephropathy.