Abstract
Inflammation, an essential cytokine-mediated process for generating a neutralising immune response against pathogens, is generally protective. However, aberrant or excessive production of proinflammatory cytokines is associated with uncontrolled local and systemic inflammation, resulting in cell death and often irreversible tissue damage. Uncontrolled inflammation can manifest over timescales spanning hours to years and is primarily dependent on the triggering event. Rapid and potentially lethal increases in cytokine production, or ‘cytokine storm’, develops in hours to days, and is associated with cancer cell-based immunotherapies, such as chimeric antigen receptor T-cell therapy. On the other hand, some bacterial and viral infections with high microbial replication or highly potent antigens elicit immune responses that result in supraphysiological systemic cytokine concentrations, which manifest over days to weeks. Immune dysregulation in autoimmune diseases can lead to chronic cytokine-mediated tissue damage spanning months to years, which often occurs episodically. Upregulation of IL-1, IL-6, IFN-γ, TNF, and granulocyte macrophage colony-stimulating factor frequently coincides with cytokine storm, sepsis, and autoimmune disease. Inhibition of proinflammatory molecules via antagonist monoclonal antibodies has improved clinical outcomes, but the complexity of the underlying immune dysregulation results in high variability. Rather than a ‘one size fits all’ treatment approach, an identification of disease endotypes may permit the development of effective therapeutic strategies that address the contributors of disease progression. Here, the authors present a literature review of the cytokine-associated aetiology of acute and chronic cytokine-mediated tissue damage, describe successes and challenges in developing clinical treatments, and highlight advancements in preclinical therapeutic strategies for mitigating pathological cytokine production.
INTRODUCTION
Cytokines are essential regulators of the immune response that mediate protective inflammation, but uncontrolled production by hyperactivated immune cells induces toxicity and adverse conditions. Pathologies that arise from excessive inflammation driven by a ‘cytokine storm’ are observed in cytokine release syndrome (CRS), systemic inflammatory response syndrome (SIRS), and sepsis.1,2 The severity can vary substantially, ranging from mild symptoms to potentially life-threatening conditions. Mild symptoms are temporary and include fatigue, muscle and joint pain, headache, fever, and rash. In more severe cases, immune hyperactivation may lead to acute respiratory distress syndrome, haemophagocytic lymphohistiocytosis, disseminated intravascular coagulation, and multiorgan failure.3,4 These symptoms are driven by local and systemic hyperphysiological concentrations of one or more cytotoxic effector cytokines, which include IL-6, IL-1, granulocyte macrophage colony-stimulating factor (GM-CSF), IFN-γ and TNF.4 Innate immune cells, primarily monocytes and macrophages, as well as T cells of the adaptive immune system, are key participants and often work in concert to amplify cytokine production, which results in the characteristic symptoms (Table 1).5,6
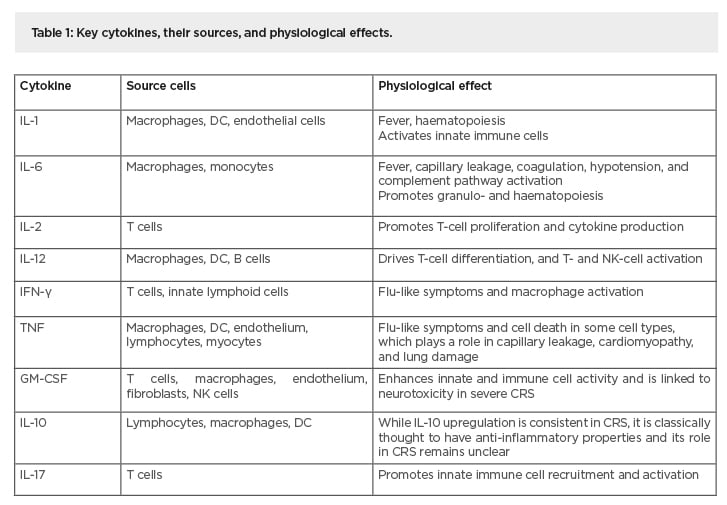
Table 1: Key cytokines, their sources, and physiological effects.
CRS: cytokine release syndrome; DC: dendritic cells; GM-CSF: granulocyte macrophage colony-stimulating factor; NK cells: natural killer cells;
Aberrant cytokine production by hyperactivated immune cells may be triggered by infections, immunotherapies, and autoimmune conditions. The manifestation of excessive cytokine production may be immediate, delayed, and/or persist as a longer-term organ- or tissue-specific chronic inflammatory condition.7 Rapid development of CRS over a few hours to days has been documented in monoclonal antibody (mAb) therapies designed to promote graft acceptance or cancer clearance, as well as post-infusion of engineered T-cell therapies (Figure 1A).8-11 On the other hand, infection by microbes that elicit a particularly intense immune response or that have a high replicative potential may result in SIRS-associated sepsis that manifests over several days to weeks (Figure 1B).12 Exemplary SIRS-like pathology is observed in some patients with acute manifestations of coronavirus disease (COVID-19), in which elevated serum IL-6 correlates with respiratory and organ failure, with adverse clinical outcomes.13 The use of immunosuppressive drugs has had limited success in managing SIRS-like pathologies. For example, the use of corticosteroids to treat inflammation arising from severe acute respiratory syndrome and Middle East respiratory syndrome did not improve mortality but delayed viral clearance.14,15 Conversely, dexamethasone treatment lowered mortality among COVID-19 patients receiving respiratory support but not among those who did not receive respiratory support, suggesting that the benefit of glucocorticoid-modulated inflammation to mitigate lung injury may be nuanced and depend on disease severity.16 Other long-term and episodic inflammation is associated with autoimmune conditions and spans weeks to years, such as that observed in rheumatoid arthritis (RA), systemic lupus erythematosus, or chronic graft-versus-host disease (cGvHD) (Figure 1C). For such conditions, broad immunosuppressive drugs increase susceptibility to opportunistic infections.
Common features associated with the hyperproduction of cytokines permit the development of therapies that might be applicable across different forms of CRS that have similar aetiology. A widely used strategy is to block the activity of the cytokines or their cognate receptors, an approach with origins in the management of rheumatic disease. As a participant in RA, TNF was the first cytokine to be fully validated as a therapeutic target.17
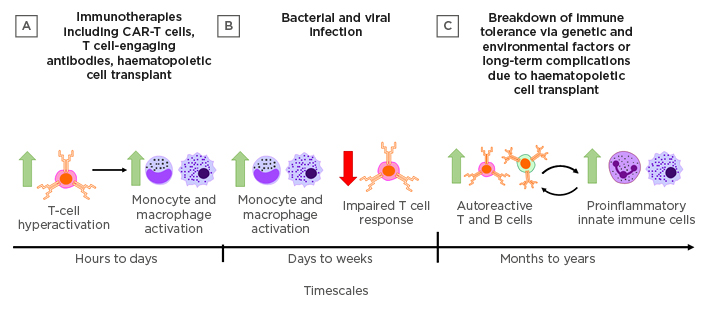
Figure 1: Triggers and timescales in the development of cytokine-mediated tissue damage.
Cytokine-mediated tissue damage may manifest in different forms across a range of timescales depending on the triggering events. A) Immunotherapies, including CAR-T cells, T cell-engaging antibodies, and haematopoietic cell transplant can all drive rapid T-cell hyperactivation and subsequent activation of innate immune cells that may result in cytokine release syndrome within hours. B) Microbial infections with high replicative potential or particularly virulent antigens may result in hyperactivation of innate immune cells over days to weeks. Delayed viral clearance leading to sepsis may also be associated with impaired T cell responses. C) Genetic factors and environmental triggers can combine to result in the breakdown of immune tolerance mechanisms, leading to chronic autoimmune disease in which autoreactive adaptive immune cells mediate cyclic cytokine-driven inflammation and tissue damage.
CAR-T cells: chimeric antigen receptor T cells.
Clinical trials using a combination of mAb targeting TNF (adalimumab) and methotrexate have an established record of safety and clinical efficacy for effectively reducing cytokine-mediated tissue damage in RA.18 However, in RA refractory to TNF inhibition, alternative therapeutic targets are necessary to induce remission. IL-6 is a participant in both RA and more acute forms of CRS, and the administration of mAb against the IL-6 receptor (tocilizumab) is frequently used to treat RA that is refractory to methotrexate or TNF inhibition.19 Tocilizumab is also clinically approved by the U.S. Food and Drug Administration (FDA) for the treatment of chimeric antigen receptor T (CAR-T) cell-induced CRS, with proven efficacy, minimal side effects, and without negatively affecting response to therapy.20 More recent developments in RA focus on inhibiting GM-CSF using mavrilimumab, an inhibitor of GM-CSF receptor α. In clinical Phase II RA trials, GM-CSF antagonism has efficacy similar to that of TNF blockade in mitigating tissue destruction.21 Early clinical data from GM-CSF inhibition in patients with COVID-19 pneumonia suggests potential efficacy in improving clinical outcomes, and a follow-up of randomised controlled trial is underway (COMBAT-19).22,23 Siltuximab, an FDA-approved IL-6 agonist for use in multicentric Castleman disease, is undergoing Phase III clinical trials for the treatment COVID-19-associated immune hyperactivation.24,25 However, the complete cytokine profile of CRS is diverse, involving the monocyte and macrophage-associated cytokines IL-8, IL-10, IL-12, TNF, IFN-α, monocyte chemotactic protein-1 and macrophage inflammatory protein-1α, in addition to the aforementioned cytokines.26 Therefore, while targeted cytokine inhibitors mitigate aberrant cytokine-mediated tissue destruction, the effectiveness of cytokine-targeted therapeutics is difficult to predict because of the complex network of cytokines and
disease heterogeneity.
This review discusses recent work that has advanced the understanding of cytokine-mediated tissue damage with distinct onset profiles arising from cancer immunotherapy, infection, and autoimmune disease. The authors describe the contributions of cytokines in disease pathogenesis, and how existing mAb therapies are repurposed to treat cytokine-mediated tissue damage. Lastly, the authors describe recent preclinical work developing new therapeutic options to mitigate damage from hyperactivated immune cells.
CYTOKINE-MEDIATED TISSUE DAMAGE ON SHORT TIMESCALES
Immunotherapies
Acute cytokine release is associated with cell-based cancer therapies such as engineered T cells, and T cell-activating immunotherapies. It is well recognised that abrogating cytokine-mediated off-target toxicity is a critical step in their widespread application. These therapies derive their efficacy, in part, from nonphysiologic T-cell activation that permits rapid and sustained production of effector cytokines. While such behavior is programmed to promote antitumour efficacy, it also leads to the unintentional consequence of notable toxicity in some cases, which typically develops within a few days after infusion and, if left untreated, may lead to death. Serum IL-6, IL-10, and IFN-γ are among the core cytokines that are consistently found to be elevated in serum CRS, which is initiated by the release of IFN-γ by activated T cells or the tumour cells.4 Generally, a higher tumour burden at the time of infusion and a greater peak in the expansion of CAR-T cells increases the risk of severe CRS. Conversely, an improved clinical outcome is not predicated on the development of severe CRS, and an effective antitumour response may be induced in the absence of this toxicity.27
While initiated by T cell-produced cytokines, CRS in CAR-T cell therapy is also dependent on the engagement of the innate immune system.6 CAR-T cell-produced IFN-γ and GM-CSF activate macrophages and monocytes, resulting in upregulation of IL-1 and IL-6 signaling. In a murine leukaemia model treated with CAR-T cells infused intraperitoneally, it was observed that macrophages in the peritoneal cavity, and not the spleen, had upregulated activation, suggesting that localised macrophage signaling plays a key role in the pathogenesis of CRS.6 Because of the involvement of macrophages, the inducible nitric oxide synthase, an enzyme indicative of macrophage activation, has been identified as a potential biomarker of CRS, in addition to IL-1 and IL-6. Current therapeutic options for CAR-T cell-associated CRS involve the blocking of inflammatory cytokine-mediated signalling. IL-6 receptor (IL-6R) blockade has generally been accepted as the front-line treatment for CAR-T-mediated CRS. The IL-6R blocker tocilizumab has been shown to reverse CRS in some patients, though some patients manifest tocilizumab-refractory CRS.28 Furthermore, early clinical results suggest that successful ablation of CRS symptoms with tocilizumab may not be sufficient to prevent delayed neurotoxicity.29,30 The pathology of CAR-T cell-induced CRS was studied in a humanised mouse model, and the upregulation of IL-1 preceded IL-6 upregulation. Treatment with an IL-1 receptor antagonist (anakinra) successfully inhibited both short-term CRS-mediated tissue damage and long-term neurotoxicity, while tocilizumab only mitigated short-term CRS, indicating that IL-1 signalling may be the initiator of CRS.31 This was further corroborated by successful treatment of CRS-like pathology in mice by inhibiting IL-1 signalling via an IL-1 receptor antagonist.6
While high IL-6 and IFN-γ are associated with CRS in CAR-T cell-based therapy, CRS may also manifest at lower levels of IL-6 and IFN-γ, and instead present as a higher concentration of IL-2 and GM-CSF.32 Because IL-2 is necessary for CAR-T cell activity, approaches that inhibit GM-CSF may be preferable to mitigate this form of CRS.33 Ibrutinib, a drug which inhibits the IL-2-induced tyrosine kinase activity, has been shown to reduce serum cytokine levels in mice.34 GM-CSF neutralisation with lenzilumab prevents CRS and neuroinflammation in mouse models of acute lymphoblastic leukaemia. Additionally, mice treated with lenzilumab and CD19-targeted CAR-T cells had enhanced antitumour efficacy compared to those treated with CAR-T cell therapy alone. When engineered with a GM-CSF knockout gene, CAR-T cells had enhanced survival rates and tumour control. These results suggest that among the inflammatory milieu, GM-CSF may be one of the crucial mediators of CRS-associated complications and could be an effective therapeutic target for controlling CRS.35
CYTOKINE-MEDIATED TISSUE DAMAGE ON INTERMEDIATE TIMESCALES
Viral and Bacterial Infections
Excessive cytokine production is associated with complex interactions between bacterial or viral pathogens and the host, inducing hyperactivation of immune cells.36 The immune response is typically characterised by an initial intense inflammatory response that rapidly peaks to increase local coagulation and thereby restrict tissue damage.37 However, an overwhelming production of these proinflammatory cytokines can result in infection-induced SIRS, termed sepsis; disrupt regulation of the immune response; and induce pathological inflammatory disorders, such as capillary leakage, tissue injury, and lethal organ failure. Generally, a high infection burden, superantigens, virulence factors, resistance to opsonisation and phagocytosis, and antibiotic resistance lead to sepsis progression when the host cannot inhibit the infection. In addition, serum concentrations of the anti-inflammatory cytokine IL-10 have been shown to parallel the sepsis score, and a high IL-10:TNF-α ratio is a predictor of severity and fatal outcomes in sepsis.38
In contrast to CRS in T cell-engaging therapies, sepsis caused by persistent bacterial and viral infection develops primarily via the innate immune system.39 Innate immune cells are activated upon recognition of exogenous and endogenous pathogen-associated molecular patterns. High microbial replication may induce hyperactivation of innate immune cells, and additional off-target tissue destruction may result in a positive feedback loop of tissue destruction and immune activation. E.g., in H5N1 human influenza A, high viral load and excess cytokine production were associated with fatal outcomes, even with low T cell counts.40 In addition, genetic polymorphisms contribute to excess cytokine production and impaired resolution of inflammation, and contribute to the variability in sepsis pathogenesis. A single nucleotide polymorphism on the IL-1 receptor antagonist gene is associated with lower plasma levels of IL-1β and improved sepsis survival, while specific alleles of the TLR4 and TLR1 genes have negative impacts on sepsis outcomes due to enhanced signaling and cytokine production.41,42 While these and several other polymorphisms relating cytokine signalling to sepsis and severe infection outcomes have been identified, clinical trials targeting individual cytokine pathways related to identified polymorphisms have demonstrated limited efficacy.39
Colonisation of barrier tissues by bacterial biofilms elicits an immune response that may lead to localised and potentially systemic cytokine-mediated tissue damage, of which periodontitis (PD) is exemplary. Although periodontopathic bacteria are the aetiological agents in PD, the primary determinant of disease progression and clinical outcome is the host immune response, which involves the generation of cytokines, and recruitment of inflammatory cells.43 While the ideal outcome of inflammation is resolution, uncontrolled inflammation, mediated by IL-17, TNF, IL-6, and IL-1 in PD, upregulates matrix metalloproteinases and the receptor activator of NF-κB ligands (the primary activation factor for osteoclasts), leading to tissue injury and scarring, fibrosis, alveolar bone destruction, and tooth loss.44-47 Surgical intervention may be necessary in severe forms of PD. However, in the face of uncontrolled inflammation, reconstruction of periodontal tissues is significantly hampered.48-50 Furthermore, chronic PD may lead to increased systemic inflammation either via locally produced cytokines entering systemic circulation, or via translocation of pathogenic bacteria to lung or heart tissues from the initial gingival ulcers.51
Severe and often lethal clinical outcomes of COVID-19 infection are associated with CRS-like symptoms that affect multiple organs, including the lungs. In a murine model of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), a delayed hyperproduction of IFN-β was linked to high levels of inflammatory monocyte-macrophage (IMM) infiltration to the lungs, resulting in mortality.52 Early administration of intranasal IFN-β or depletion of IMM improved survival and T-cell response. Early clinical results testing IFN-βfor the treatment of severe COVID-19 suggest a lower 28-day mortality, consistent with results from the murine model of SARS-CoV.53 Delayed hyperproduction of IFN-β also induced T-cell lymphopenia in mice, which may further contribute to increased viral load and IMM hyperactivation, as virus-specific T cells are required for viral clearance.54,55 Similar to cancer immunotherapy-associated CRS, high levels of IL-1 and IL-6 were correlated with disease severity. However, while early results suggested that using tocilizumab alone to target IL-6R might be an effective therapeutic target to suppress hyperactive inflammation in CRS, a recently concluded Phase III clinical trial (COVACTA)56 did not meet its primary endpoint of improved clinical status in patients with COVID-19-associated pneumonia, or the key secondary endpoint of reduced patient mortality, underscoring the complexity of the cytokine network underlying the disease.57 Results from ongoing clinical trials testing single-target cytokine inhibition for the treatment of severe COVID-19 are awaited, with additional testing planned for antibody cocktails targeting one or more of IL-6, IL-1, TNF, and GM-CSF.58
CYTOKINE-MEDIATED TISSUE DAMAGE ON EXTENDED TIMESCALES
Autoimmune Diseases and Autoimmune-like Conditions
In autoimmune diseases and autoimmune-like conditions, immune dysregulation contributes to episodic elevation in cytokine production and chronic inflammation that may last over a lifetime. In contrast to acute CRS, which is associated with multiorgan failure, damage from autoimmune diseases are primarily tissue-specific, and chronic conditions contribute to long-term collateral damage of organs such as the skin, eyes, lungs, and heart. Proinflammatory cytokines drive pathogenesis in RA as well as systemic lupus erythematosus, and flares of disease activity are associated with increased cytokine production.59Comprehensive efforts have developed a high resolution map of the hierarchical position of distinct cytokines, which mediate the overlapping innate and adaptive immune responses associated with disease onset and persistence in RA pathogenesis.60 Preclinical and clinical studies, along with the success of cytokine-targeting drugs, such as anti-TNF and anti-IL-6R, have validated the pivotal contribution of cytokines in the pathogenesis of RA. In RA, cytokines regulate cellular phenotype, localisation, activation status, and longevity in the synovial and lymphoid microenvironments, supporting a role for cytokines in the licensing of cell function in RA rather than simply as strict differentiation factors. However, in contrast to a specific antigen or pathogen, the immune response in RA is not thought to be synchronised by a specific initiating event; therefore, the usual innate and adaptive cellular responses are unlikely to operate in the rheumatoid joint. The net effect of this cellular profile is the generation of tissue-destructive enzymes, reactive oxygen and nitrogen intermediates, prostaglandins and leukotrienes, and a broad range of effector cytokines, outside their normal homeostatic ‘on–off’ regulatory cycle, often following an unpredictable schedule. The result is that the therapy needs for each patient may be distinct and an ad hoc combination is often employed to induce disease remission.
In cGvHD, cytokine production by autoreactive donor T cells drives immunological dysregulation and tissue damage. IL-17-producing T helper (Th17) cells are thought to drive the pathogenesis of cGvHD, and targeting the Th17 axis has been shown to ameliorate cGvHD in preclinical models.61,62 A mAb targeting the p40 subunit found on both IL-12 and IL-23 reduced the production of both IFN-γ and IL-17 and reduced tissue damage in the skin and salivary glands in a preclinical model of cGvHD.61 In a retrospective analysis, tocilizumab has shown potential in treating cGvHD, as IL-6 signalling is necessary for Th17 differentiation; however, more comprehensive clinical studies may be necessary to establish clinical efficacy.63 Furthermore, the contributions of cytokines in cGvHD is complex and may be source-dependent. Host and donor cytokines may play opposing roles, and cytokines may be protective in some tissues but damaging in others, which may drive the selection of which cytokines to suppress. For example, recipient IL-22 has demonstrated protectivity for intestinal stem cells in cGvHD, whereas donor-derived IL-22 plays a critical role in driving cutaneous cGvHD.64 Therefore, while broadly targeting IL-22 may alleviate cGVHD symptoms, it may not represent the optimal cytokine for inducing disease remission.
To date, there is no single successful strategy to manage cGvHD in the clinic. The clinical gold standard of using combination cyclosporin and methotrexate, which has remained unchanged for decades, is only partially effective.65 In addition to lengthening the period of immune deficiency, in the order of several years or even the lifetime of the individual, some patients may develop steroid-resistant cGvHD. A strategy that has demonstrated promise in controlling cGvHD is the selective expansion of Tregs, mediated by systemic IL-2 infusions.66,67 However, in clinical trials daily injections were needed for therapy, and patients experienced symptoms of cGvHD immediately following cessation of treatment. mAb, such as the anti-CD20 antibody rituximab, have been shown to be useful in several clinical trials for the treatment of cGVHD; however, these therapies are administered over the lifetime of the patients, significantly impacting the immuno-competence of an individual and thereby limiting its applicability.
INNOVATIVE TREATMENTS IN THE CLINICAL AND PRECLINICAL PIPELINE
While current frontline mAb treatments for specific cytokines are effective at mitigating CRS in some patients, targeting multiple pathways may improve efficacy and applicability (Figure 2A). One method employed is broad-spectrum cytokine absorption with biomimetic nanoparticles to reduce undesired cytokine signalling (Figure 2B). Nanoparticles coated with neutrophil membrane have been shown to reduce proarthritogenic factors such as IL-1β, TNF-α, and matrix metalloproteinase-3, and ameliorate experimental arthritis in both a collagen-induced arthritis model as well as in a TNF-transgenic mouse.68 Dendrimers, which are highly branched macromolecules with polyvalent adsorption capabilities, have been demonstrated to mitigate adverse cytokine-mediated tissue damage. In a rhesus macaque model of Shigella dysenteriaeinfection, orally administered dendrimer glucosamine significantly reduced colonic levels of IFN-γ, IL-1β, IL-6, and IL-8, and conferred protection against neutrophil-mediated vasculitis and gut wall necrosis.69 A hydroxy dendrimer, termed OP-101, has been shown to inhibit multiple macrophage cytokine pathways and is currently undergoing Phase II clinical trials for the treatment of patients with severe COVID-19 (PRANA).70Such nanoparticle medicines have the advantage of being able to simultaneously target multiple pathways while still providing rapid clearance and off-the-shelf convenience.
Alternatively, directly targeting cells affected by CRS may prove useful to reduce tissue damage (Figure 2C). Recent multiomics profiling of endothelial, epithelial, and stromal cells demonstrated that these structural cells play critical roles in immune regulation in a tissue-dependent manner.71 In a murine model of influenza infection, agonism of sphingosine-phosphate-1 reduced chemokine production by pulmonary endothelial cells and mortality due to cytokine storm.72 Additionally, the Slit2/Robo4 signalling in endothelial cells may be a therapeutic target, as Slit2 inhibits ICAM-1 expression on endothelial cells that promote monocyte adhesion, as well as reducing lipopolysaccharide-induced production of proinflammatory cytokines by endothelial cells.73
In addition to treatments for acute manifestation of cytokine-mediated tissue damage, several treatments for chronic conditions are currently being explored (Figure 2D). Preclinical models of tolerogenic vaccination to enhance regulatory immune cell subsets and promote antigen-specific tolerance show promise and have been reviewed extensively elsewhere.74,75
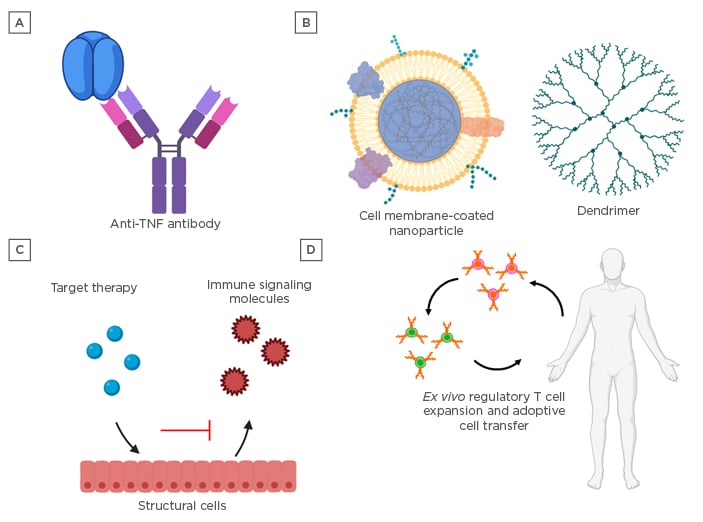
Figure 2: Treatments for cytokine-mediated tissue damage.
A) Monoclonal antibodies and broad-spectrum immunosuppressive molecules, such as steroidal anti-inflammatory medications, are current front-line therapeutics for the mitigation of cytokine-mediated tissue damage. B) Nanomedicines offer the advantage of targeting multiple pathways via a single platform and may be engineered with enhanced targeting capabilities to minimise off-target immune suppression. C) Advanced omic profiling has shed light on the involvement of structural cells, such as endothelial, epithelial, and stromal cells, in orchestrating immune responses, and preclinical therapies targeting these cell types have shown promise in mitigating cytokine-mediated tissue damage. D) Immunomodulatory tolerogenic vaccines and cell therapies to enhance the number and function of regulatory immune cells may hold the key to restoring immune homeostasis in chronic diseases with complex underlying cytokine networks, such as chronic graft-versus-host disease and autoimmune diseases.
Cell-based therapies to restore immune homeostasis in autoimmune disease have shown great promise.76-80Clinical trials evaluating the efficacy of transfusion of autologous tolerogenic dendritic cells and regulatory T cells are underway for Type 1 diabetes mellitus, as well as RA.78,81 While clinical outcomes and efficacy are awaited, the prospect of cell-based therapy, combined with front-line therapies targeting cytokines for debilitating chronic inflammatory disease, may be a promising strategy for patient-specific long-term disease remission.
CONCLUSION
Cytokine-mediated inflammation is an essential component of the natural course of an immune response. However, immunological dysregulations that arise from immunotherapies, persistent or highly immunogenic microbial infection, and underlying genetic predisposition, can result in the supraphysiological production of cytokines that results in harmful toxicity. Common features associated with the hyperproduction of cytokines have resulted in therapies that are transferable and effectively manage symptoms, independent of disease aetiology. Current treatments for these pathologies using mAb to inhibit key inflammatory cytokines have shown promise, but patient-to-patient response can be highly variable, in part because of the complex underlying cytokine network. Therefore, therapies targeting multiple pathways may improve outcomes and management of CRS. Identifying the key cellular and molecular determinants of immune tolerance and their role in immune dysregulation will characterise differences between distinct manifestations of CRS, as well as classify patient subsets and better predict therapeutic targets.






