
Abstract
Early diagnosis and personalised disease management remain serious challenges in the field of haematological malignancies, especially for acute conditions. Additionally, reliable and timely detection of minimal residual disease is the key to improving patients’ clinical management, including guiding therapy choice, monitoring treatment response, and detecting relapse. Genomic biomarkers provide valuable information in this regard.
For example, chromosomal translocations and mutations in genes involved in haematopoietic proliferation and differentiation are considered the strongest predictors of treatment response and overall survival in acute myeloid leukaemia. Furthermore, NPM1 gene mutations and internal tandem duplications in FMS-like tyrosine kinase-3 (FLT3) are used to monitor minimal residual disease in acute myeloid leukaemia. Despite the growing availability of relevant molecular biomarkers, long turnaround times for genomic testing greatly impact the management of aggressive haematological disorders. Delayed access to laboratory test results both negatively influences patients’ psychological state and postpones therapy administration and adjustments. Depending on the technology used, next-generation sequencing allows high-throughput genome analyses within hours to days, at a relatively low cost. Simultaneously, it enables testing of large numbers and various types of biomarkers by targeted gene panels. At the present time, with the latest technological improvements, next-generation sequencing provides the means for advancing genomic-based diagnostics in haematological malignancies, by simplifying complex laboratory workflow and should be introduced more widely in routine clinical settings.
BACKGROUND
Haematological malignancies (leukaemia, lymphomas, and myelomas) comprise a heterogeneous group of more than 60 subtypes of cancer, many of which have unique clinical pathways and outcomes.1 When compared to other cancers, the path to their diagnosis can be difficult and challenging. Specifically, the time between symptom onset, seeking medical aid (from primary to secondary care), and establishment of a reliable diagnosis can span several months. Furthermore, the clinical presentation of haematological malignancies is often broad and relatively poorly defined. This is particularly true for the initial symptoms which are usually nonspecific, associated with a long prodromal period, and are difficult to discriminate from those of benign tumours. Therefore, early diagnosis of haematological malignancies remains a challenge to overcome in addition to representing an important determining factor for a patient’s outcome.2 Kariyawasan et al.3 previously found that diagnostic delay contributes to increased complications in patients with multiple myeloma.3
Besides optimising the time intervals leading to secondary care, shortening the diagnostic period can improve downstream clinical management of patients. Therefore, the development of reliable and specific molecular diagnostic methods that offer fast turnaround times (TAT) are necessary. This will not only facilitate personalised treatment allocation, but additionally could allow monitoring and management of disease progression. In addition, routine testing of clinical samples of haematological malignancies will contribute to the proliferation of a real-world sample database and characterisation of novel genetic abnormalities, that in turn could trigger the development of a wider range of targeted treatment options.
This review centres on the impact next-generation sequencing (NGS) can have on the current management of haemato-oncological diseases, with a special focus on comprehensiveness and speed of current molecular test strategies.
SUCCESSFUL TALES OF TARGETED DRUG THERAPIES ARE BACKED BY APPROPRIATE GENOMIC TESTING
Acute Myeloid Leukaemia
Acute myeloid leukaemia (AML) is a highly heterogeneous disease that results from large chromosomal translocations, as well as mutations in genes involved in haematopoietic proliferation and differentiation. The standard therapy remains induction chemotherapy with a combination of cytarabine and an anthracycline, and allogeneic stem cell transplantation for suitable patients. With the rise of new treatment options, the genetic complexity of AML poses a great challenge for diagnosis and disease management.4 Overall, the opportunity to comprehensively address multiple genomic aberrations at once will be pivotal for this condition.
Clonal chromosome alterations are found in more than 50% of adult AML cases and represent the strongest predictors of response duration, treatment resistance, and overall survival.5 According to the 2017 European LeukaemiaNet (ELN)6 from an international expert panel, AML risk assessment depends on the presence of fusion transcripts as well as specific gene mutations (Figure 1). Among these are t(6;9), inv(3)/t(3;3), t(v;11q23.3), and t(9;22) abnormalities, which are confirmed predictors of poor outcome,6 and mutations in NPM1, TP53, RUNX1, and ASXL1 genes. Immediate therapy initiation was traditionally deemed crucial to minimise disease-related morbidity and mortality;7 however, in some cases, prognostic classification is currently performed before AML treatment to spare unnecessary, aggressive chemotherapy for low-risk, stable patients.8
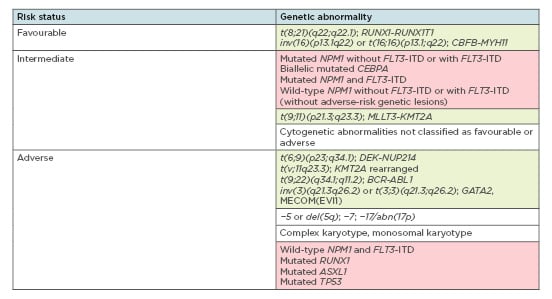
Figure 1: Overview of genetic abnormalities used for risk stratification of acute myeloid lymphoma patients according to the 2017 ELN recommendations.6
Green boxes: chromosomal aberrations; red boxes: gene mutations.
Adapted from Döhner et al.6
Recent advancements in the genetic characterisation of AML led to the identification of additional recurrent genetic mutations, such as internal tandem duplication of the FLT3 gene and IDH. Examples of U.S. Food and Drug Administration (FDA)-approved targeted drugs for use in AML include the multikinase inhibitors midostaurin (Rydapt®, Novartis, Basel, Switzerland) and gilteritinib (Xospata®, Astellas Pharma, Tokyo, Japan) for FLT3 mutation-positive patients, and the IDH inhibitors ivosidenib (Tibsovo®, Agios Pharmaceuticals, Cambridge, Massachusetts, USA) and enasidenib (Idhifa®, Celgene, Summit, New Jersey, USA) for adult patients with relapsed or refractory disease with an IDH1 or IDH2 mutation, respectively. Testing for FLT3, IDH1, and IDH2 mutations is now mandatory for haematologists attributable to the availability of these drugs and their clinical relevance.9
TAT in mutation status testing and its impact on the management of clinically aggressive AML is an extremely important point to be considered.10 The heterogeneous nature of genetic aberrations in AML, which include both DNA mutations and RNA fusions, ideally require a molecular method that can assay both types of markers. At present, all clinically relevant fusions can be detected using a RNA-based NGS assay.11 Acute promyelocytic leukaemia PML-RARA has to be tested within 24 hours and is the strongest exponent for fast TAT. Commercially available panels can be used to profile DNA mutations, for example, the Illumina® (San Diego, California, USA) TruSight Myeloid Sequencing Panel, the ArcherD® (Boulder, Colorado, USA) VariantPlex® Core Myeloid panel, and the Human Myeloid Neoplasms QIASeq® Targeted DNA Panel (Qiagen, Hilden, Germany). Other panels can profile DNA mutations and RNA fusions simultaneously, such as the AmpliSeq for Illumina® Myeloid panel and the Oncomine™ Myeloid Research Assay (Thermo Fisher, Waltham, Massachusetts, USA), providing a more comprehensive testing solution. Given the complexity and the number of molecular biomarkers to be tested, NGS is currently the most cost-effective method for determining the mutational status of genes for AML
risk assessment.
Acute Lymphocytic Leukaemia
Acute lymphocytic leukaemia (ALL) originates from the malignant transformation of B and T lymphoid precursor cells, referred to as B-cell ALL and T-cell ALL, respectively. It is caused by several genetic abnormalities including chromosomal translocations, mutations, and aneuploidies.12,13 Notably, the biological characteristics of paediatric ALL differ from those of the adults, and the latter are associated with poorer outcomes. For this reason, ad hoc paediatric cancer-focussed diagnostic solutions are urgently needed. The recent introduction of the Oncomine Childhood Cancer Research Assay and AmpliSeq™ for Illumina Childhood Cancer Panel represents a major step forward towards addressing this need, thus partially mitigating this issue. Nonetheless, regarding treatment, multiagent chemotherapy is the first-line therapeutic approach for both paediatric and adult ALL, followed by haematopoietic stem cell transplantation in high-risk patient groups.14,15
THE PRESENCE OF THE PHILADELPHIA CHROMOSOME, t(9;22) TRANSLOCATION
Minimal residual disease (MRD) is the key cytogenetic hallmark in Philadelphia-positive ALL (Ph-positive ALL), with the greatest impact on disease prognosis and treatment.14 In more than 80% of cases, the variant has deletions in key transcription factors involved in B-cell development including IKAROS family zinc finger 1 (IKZF1), transcription factor 3 (E2A), early B-cell factor 1 (EBF1), and paired box 5 (PAX5).16 Other genetic variations used for ALL risk assessment and targeted therapy include deletion of IKZF1 that is associated with poor outcomes,17-21 high rates of remission-induction failure, as well as relapse in patients with B-cell ALL,22 and deletions of ERG, CDKN2A/B, PAX5, EBF1, RB1, and ETV6 that negatively influence prognosis.23-28 Table 112 summarises an overview of currently known key genetic alterations in ALL and their clinical significance.
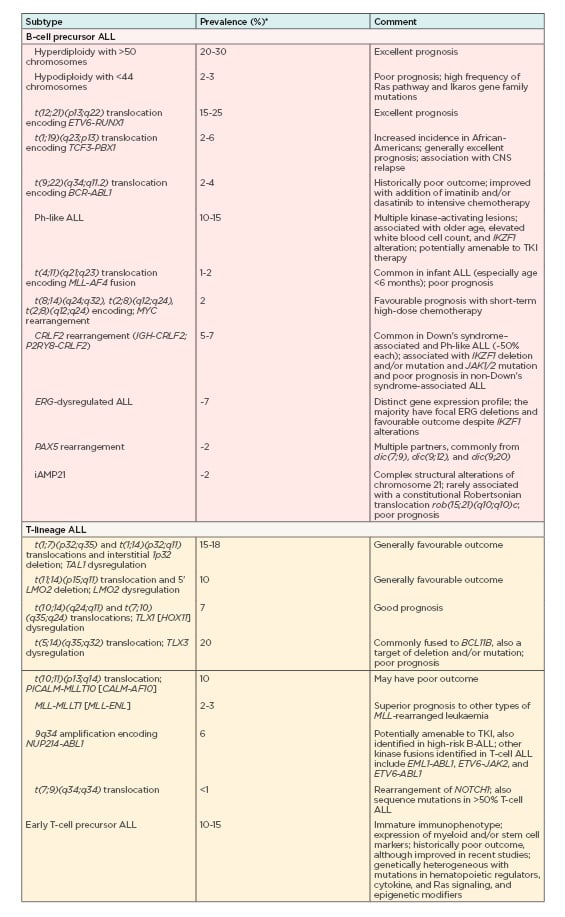
Table 1: Overview of currently known key genetic alterations in acute lymphocytic leukaemia, their prevalence, and their clinical significance.
ALL: acute lymphocytic leukaemia; CNS: central nervous system.
*Frequencies/prevalence refer to childhood ALL.
Adapted from Hunger et al. (2015)12
Patients with kinase-activating mutations comprise 90% of Philadelphia-like ALL cases and are treated with ALK kinase inhibitors ruxolitinib (Jakafi®, Incyte, Wilmington, Delaware, USA), and crizotinib (Xalkori®, Pfizer, New York, New York, USA). However, more recently monoclonal antibodies, immunomodulators, and chimeric antigen receptor T cells have been successfully introduced as additional therapy options.29 Examples of FDA-approved monoclonal antibodies for the treatment of refractory and relapsed adult ALL include blinatumomab (Blincyto®, Amgen, Thousand Oaks, California, USA) and inotuzumab ozogamicin (Besponsa®, Pfizer) where the latter is combined with an anticancer agent. For the treatment of ALL in children and young adults, chimeric antigen receptor T cells have recently been approved and calaspargase pegol-mknl (Asparlas®, Servier Pharmaceuticals), an asparagine-specific enzyme, has been added into the multi-agent chemotherapeutic regimen. As for AML, considering the complexity and the number of molecular biomarkers to be tested, NGS is increasingly being used for routine testing either as a replacement of, or along with, cytogenetics analysis, fluorescence in situ hybridisation, PCR, Sanger sequencing, reverse transcription-PCR, and multiplex ligation-dependent probe amplification.13
APPROACHES TO DETECT MINIMAL RESIDUAL DISEASE
MRD is a powerful prognostic parameter in the clinical management of haematological malignancies. It is defined by the presence of residual leukaemic cells in the blood circulation (Figure 2).30 MRD assessment can help allocate patient risk groups, assist therapy choice, as well as predict progression-free survival and overall survival.
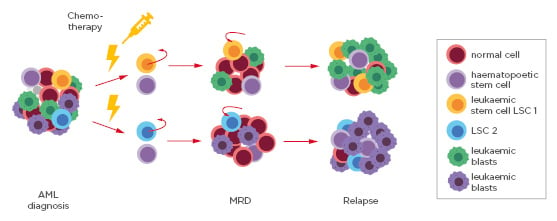
Figure 2: Schematic of the sequential steps from acute myeloid leukaemia diagnosis, to minimal residual disease detection and relapse.
The pool of blood cells at diagnosis consists of normal cells, haematopoietic stems cells, leukaemic blasts, and different leukaemic stem cells (LSC1 and LSC2). Following standard chemotherapy and radiotherapy treatment, the bulk of the cells die except for residual LSC that mark MRD. LSC can replicate and ultimately lead to relapse.
AML: acute myeloid leukaemiaLSC: leukaemic stem cells; MRD: minimal residual disease.
Multiple genomic biomarkers can be used to monitor MRD in haematological malignancies, thus making NGS an attractive solution to address them all at once.31 Somatic mutations in exon 12 of NPM1 are the most frequent genetic abnormalities in adult AML and found in approximately 35% of all cases and up to 60% of patients with normal karyotype AML.32 As NPM1 mutations show great stability during disease evolution, they represent a reliable marker for MRD detection.
Similar to NPM1, FLT3-ITD is one of the most important targets for MRD assessment in AML, as well as RUNX1 abnormalities that characterise de novo AML and associate with favourable patient prognosis, although they have to be tested in conjunction with additional recommended biomakers.13,33,34
Other targets for monitoring MRD that could be assessed by NGS in myeloid neoplasms include RUNX1-RUNX1T1 fusion gene quantification,35,36 and single nucleotide variation analysis of several genes including RUNX1, TP53, TET2, DNM3TA, and IDH1/2.37,38
Overall, MRD markers that are commonly used by all centres include NPM1 and fusion transcripts, while point mutation testing by NGS is experiencing a fast increasing adoption rate, it is in its early stages of application.
In addition, rearrangements of the IGHV gene for B-cell malignancies, or T-cell receptors for T-cell malignancies, present an appropriate MRD marker in lymphocytic disorders, considering that each malignant cell expresses the same clone of IGH or T-cell receptor. In regard to this point, besides the classical MRD monitoring by real-time quantitative PCR and flow cytometry analysis,39 novel approaches with higher precision and sensitivity have been developed. These include multi-parameter flow cytometry, allele-specific oligonucleotide qPCR, digital PCR, and NGS.40 As previously mentioned, the advantages associated with NGS include the possibility to examine multiple genomic regions at once, along with a sensible reduction of the overall laboratory workflow processes enabling the channeling of most resources into a single testing effort. To track clonality, NGS-based Ig assays are commercially available, such as the LymphoTrack® family of assays (Invivoscribe Technologies, Inc., San Diego, California, USA) or Oncomine BCR IGH SR Assay, and the Oncomine TCR Beta-SR Assay. Alternatively, custom-made solutions have been developed, as described by Onecha et al.38 where the authors established and validated a high-throughput sequencing method for determining MRD status in AML patients. This was designed to detect cell clonotypes with mutations in NPM1, IDH1/2, and/or FLT3-single nucleotide variants.38
CLINICAL SIGNIFICANCE OF TIME TO RESULTS: AN OUTLOOK ON NEXT-GENERATION SEQUENCING ADOPTION
Introducing NGS into the clinical research settings and clinical practice is of great relevance for advancing genetic-based diagnostics of different disorders, including that of haematological malignancies.41 Rapid assessment of biomarkers in haematological malignancies is relevant for making first-line therapy choices, especially where targeted therapies are available. However, having substantial technical and methodological differences, not all NGS platforms are equal. Depending on which platform is used, NGS offers high-throughput analyses within hours (e.g., the Ion Torrent Genexus System, Thermo Fisher), or days (e.g., the Illumina MiSeq, NexSeq, or the Ion Torrent S5 System, Illumina). This is done cost-effectively by simultaneously testing large numbers of genes using targeted gene panels.42 Whole-genome sequencing and whole exome sequencing allow the identification of potential novel biomarkers located within introns (for whole genome sequencing) or splice junctions, which could bear valuable prognostic information.11 While their value has been recognised in studying haematological malignancies, introduction of these methods into routine clinical testing appears to be confined to a very limited number of cases as a result of costs, long testing time to results (weeks), and data analysis and interpretation complexity. Conversely, testing via NGS targeted gene panels is already included in the 2017 European LeukemiaNet (ELN) guidelines.6 NGS can be useful in the selection of patients that meet the inclusion criteria for clinical trials and in this way contribute to the discovery of novel therapies. Using NGS to monitor MRD-positive versus MRD-negative status also helps reliably monitor treatment response, prevent disease relapse by taking timely therapeutic measures, and ultimately improve patient overall survival. Interpretation and evaluation of MRD results require close co-operation between haematologists, pathologists, and the executing laboratory. Therefore, short TAT will facilitate such an interdisciplinary effort to generate a tailored and risk-adapted therapeutic approach.43 Patients at high risk will receive a more invasive therapy, while those with a better prognosis can be spared the toxicity of a one-size-fits-all therapeutic approach.44
CONCLUSION
Novel sequencing technologies are rapidly evolving to enable rapid TAT for diagnosis, prognosis, and direction of therapy choices in myeloid disorders. Fast NGS methods are changing the current landscape of molecular diagnostics and will greatly impact the overall clinical management of patients with haematological disorders.45





