Abstract
Peripheral T-cell lymphomas are aggressive lymphomas with a rapidly progressive clinical course and sinister prognosis even with the best available treatment modalities. Epstein–Barr virus-positive peripheral T-cell lymphoma is an unusual variant of the disease; it is extremely rare and associated with a fulminant course spanning weeks to months. Treatment protocols are different for this entity because of its rarity.
INTRODUCTION
The World Health Organization (WHO) has defined peripheral T-cell lymphomas (PTCL) as a heterogenous group that is diagnosed based on typical histological findings: medium-sized or large cells with irregular, pleomorphic nuclei; prominent nucleoli and many mitotic figures; an aberrant immunohistochemical profile with the loss of CD5 and CD7, along with a clonal T-cell receptor gene rearrangement. It is associated with poor prognosis with a rapidly progressive clinical course. Epstein–Barr virus (EBV)-positive (+) PTCL is an uncommon variant with worse prognosis than PTCL not otherwise specified.1 In low-grade B-cell lymphomas, Reed Sternberg (RS)-like cells can be seen in follicular lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, and marginal zone lymphoma. Among the T-cell lymphomas, RS-like cells are seen in angioimmunoblastic lymphoma and peripheral T-cell lymphoma.2,3
CASE REPORT
A 27-year-old female presented with history of fever, weight loss, and persistent, enlarged neck swellings for 3 months. She had been diagnosed with HIV 4 years previously, and with tuberculous lymphadenitis 2 months prior to this study and started on antitubercular therapy (ATT); however, she stopped taking ATT after 1 month because of persistent vomiting and yellowish discolouration, potentially owing to ATT-induced hepatitis.
Clinical examination revealed multiple cervical and axillary lymphadenopathy ranging in size from 1.5 to 3.0 cm. The bilateral supraclavicular nodes were also enlarged. On abdominal examination, gross ascites with hepatosplenomegaly was noted. Haematological examination showed pancytopenia with a macrocytic blood picture. Ultrasonography of the abdomen was suggestive of ascites with hepatosplenomegaly and retroperitoneal lymphadenopathy.
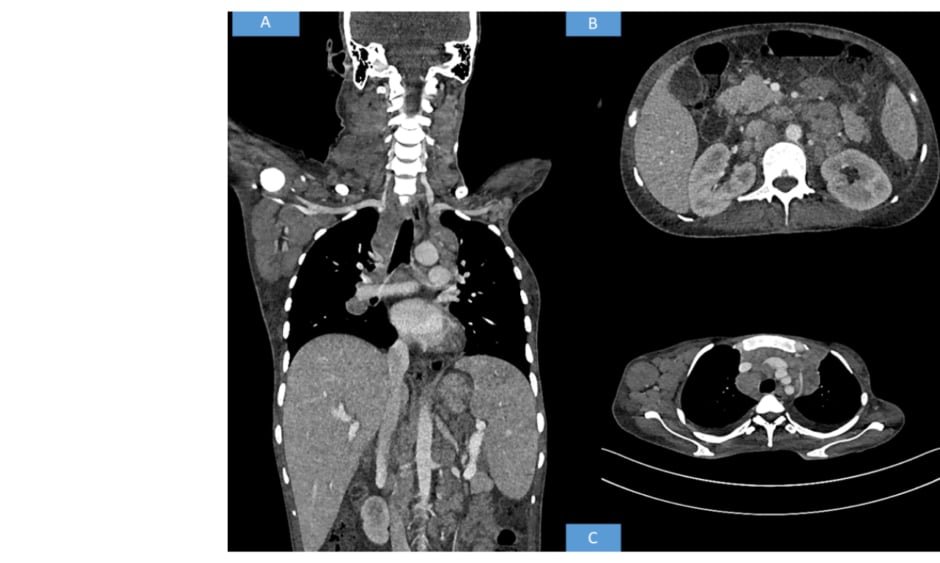
Contrast-enhanced CT of the neck, thorax, abdomen, and pelvis was advised. The CT revealed multiple discrete homogenously enhancing enlarged lymph nodes seen in cervical stations with conglomeration in few areas involving right Level II, right Level III, right Level IV, right axillary, and bilateral supraclavicular nodes. Mediastinal lymph nodes were also enlarged with bilateral pleural effusion. The enlarged preaortic, para-aortic, aortocaval, retroperitoneal lymph nodes were seen encasing and lifting the abdominal aorta without any luminal narrowing. Along with gross ascites and hepatosplenomegaly, multiple hypodense splenic deposits were also noted. Based on the above radiological findings, the possibility of a lymphoma was suggested (Figure 1).
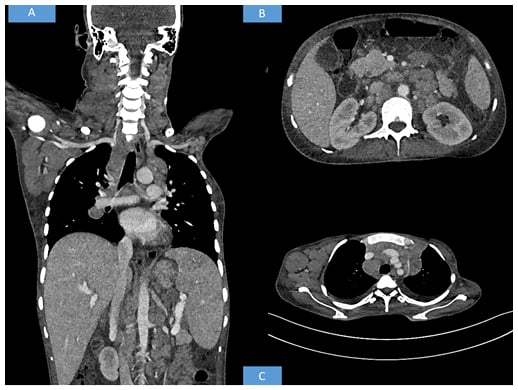
Figure 1: Radiological findings.
(A) Whole body CT shows hepatosplenomegaly with axillary and cervical lymphadenopathy. (B) CT shows retroperitoneal enlarged lymph nodes. (C) CT shows axillary and mediastinal lymphadenopathy.
Ultrasonography-guided ascitic tap was performed and sent for cartridge based nucleic acid amplification testing which gave a negative result. A lymph node biopsy was done from right Level II lymph node and sent for histopathological examination and cartridge based nucleic acid amplification testing which, again, was negative. Haematoxylin-eosin stained sections revealed diffuse effacement of the nodal architecture by small to medium-sized lymphoid cells with round or convoluted nuclei and scant cytoplasm. Interspersed between these cells were large pleomorphic cells with scant to moderate amounts of eosinophilic to vacuolated cytoplasm, large oval to irregular nuclei, vesicular chromatin, and prominent nucleoli resembling the owl eye appearance of RS cells. Based on the histological findings, the possibility of lymphocyte-depleted Hodgkin’s lymphoma and anaplastic large-cell lymphoma were considered and immunohistochemistry was ordered.
Immunohistochemistry showed that the small to medium-sized cells were positive for CD4 and CD68 while the large pleomorphic cells were only positive for EBV-latent membrane protein 1 (LMP). The bizarre cells did not show positivity for any B-cell markers (CD20, PAX5, CD79a) or for CD15, CD30, Oct-2, and Bob-1, which are positive in RS cells. HMB-45, CD1a, langerin, anaplastic lymphoma kinase, EMA, vimentin, desmin, cytokeratin, and CD56 were also tested to exclude the possibility of malignant melanoma, anaplastic large cell lymphoma, high-grade carcinoma, and natural killer cell lymphoma, and all turned out to be negative. Two differentials were concluded: EBV+ PTCL and histiocytic sarcoma. GATA Binding Protein 3 (GATA3) was positive in the large cells therefore a final diagnosis of EBV+ PTCL was made (Figure 2).
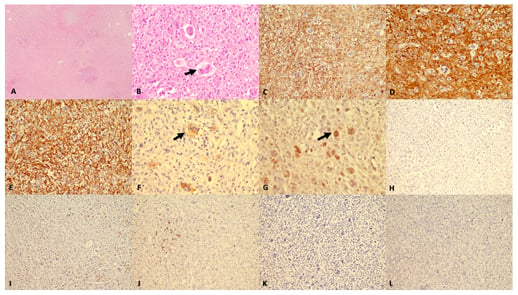
Figure 2: Histomorphology and immunohistochemistry findings.
(A) Low magnification (40x haematoxylin and eosin [H&E]) shows complete effacement of lymph node architecture with diffuse infiltration by atypical cells. (B) Higher magnification (400x H&E) shows, aside from tumour population, numerous large cells (arrow) with prominent eosinophilic nucleoli resembling Reed Sternberg cells. In background, eosinophils are also seen. The tumour cells are diffusely stained with (C) LCA; (D) CD 4; and (E) CD68. The bizarre tumour cells show strong staining (arrow) with (F) EBV LMP-1; and (G) GATA-3. The tumour cells are negative for (H) CD3; (I) CD8; (J) CD20; (K) CD30; and (L) PAX-5.
The patient was initially advised treatment with cyclophosphamide and prednisolone for lymphoma rather than full CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisolone) treatment which could not be administered because of deranged liver function. The patient was counselled regarding the decision and she agreed to the treatment; however, the patient was not reachable during the follow-up. Upon further contact it was revealed that the patient had died.
DISCUSSION
PTCL is a heterogeneous group that can have both nodal and extranodal presentation and does not fit the specifically defined entity of mature T-cell lymphoma. It usually presents in adults with a male:female ratio of 2:1. It is associated with rapid progression of clinical course.1 EBV+ PTCL, a rare variant of PTCL, is more frequent in late adulthood and has a poorer prognosis. Patients are more likely to have B symptoms and hepatic involvement. A higher stage, usually Stage III/IV, is seen in EBV+ cases.1
Histological examinations of PTCL show a mixture of small to large atypical lymphoid cells with clear cytoplasm and irregular nuclei with conspicuous nucleoli; however, EBV+ PTCL show different histomorphological findings with centroblastic, anaplastic, or plasmacytoid appearance.4 Immunohistochemical profiles of PTCL show double positive CD4+>CD8+ with aberrant phenotypes associated with loss of CD5 and CD7.5 EBV+ PTCL are CD8+/CD4-, double negative, and approximately 15% are CD4+/CD8-. Cytotoxic molecules such as TIA-1 and granzyme are expressed in nearly all cases unlike in PTCL not otherwise specified in which they may or may not be expressed.5
Immunopositivity for GATA3 has been associated with a poorer prognosis with increased infiltration with tumour-associated macrophages and higher CD68 positivity as in seen in this report.6 The pathogenesis of EBV-associated PTCL was proposed by Cheng et al.7 by two mechanisms. Firstly, EBV-associated proteins such as EBV nuclear antigen 1 or LMP transactivate certain important genes in lymphoma cells thus further stimulating the cytokine secreting T cells. Secondly, EBV may initiate an abnormal immunological reaction of host against the EBV+ T lymphoma cells.7
The aggressive course of PTCL has been attributed to a terminal haemophagocytic syndrome and rapid emergence of P-glycoprotein following a course of chemotherapy.7 Two types of haemophagocytic syndrome have been reported. Jaffe et al.8 described one in which a disease-free period was seen followed by development of terminal haemophagocytic syndrome. Bone marrow in the above subtype of syndrome showed many reactive histiocytes.8 The second type of haemophagocytic syndrome was described by Falini et al.9 in which there was simultaneous development of haemophagocytic syndrome with lymphoma followed by death after a few months. In contrast to the former, bone marrow, liver, and spleen in this syndrome showed reactive histiocytes along with lymphoma cells.9 Song et al.10 and Wang et al.11 have also reported similar cases in which, alongside generalised lymphadenopathy, erythematous papules were also present. Both cases were aged between 40 and 49 years old and immunocompetent, while the patient in this case report was in their 20s and immunocompromised. The authors’ case did not show any skin manifestations. Histomorphological findings in the above cases were similar to the case in the present report; however, on immunohistochemical analysis, the larger RS-like cells were positive for EBV-LMP and GATA3 and immunonegative for B-cell markers (CD20, CD79a). In contrast, their cases showed the large pleomorphic cells to be immunopositive for CD20, PAX-5, and CD30. They tested EBV positivity by EBV-encoded RNA in situ hybridisation while in this present case it was carried out using immunohistochemistry for EBV-LMP.
Goh et al.12 reported a similar case of a patient in their 50s with presentation of isolated inguinal lymphadenopathy. Mori et al.13 reported a case of a patient in their 70s presenting with gradual enlargement of submandibular nodes which was earlier misdiagnosed as classical Hodgkin lymphoma. The case was treated as Hodgkin lymphoma, which then recurred, only to be diagnosed as angioimmunoblastic lymphoma on a subsequent biopsy. Butler et al.14 reported a similar case of a patient in their 50s who presented with pyrexia of unknown origin. It was earlier misdiagnosed as Hodgkin lymphoma; however, a subsequent excisional biopsy revealed PTCL with RS-like cells. Willemsen et al.15 reported a case of angioimmunoblastic lymphoma with RS-like cells in a 76-year-old male patient who presented with chylothorax and chylous ascites; however, these RS-like cells were negative for EBV on EBV-encoded RNA in situ hybridisation.
CONCLUSION
The authors have reported a case of EBV+ PTCL in a 27-year-old patient. The importance of diagnosing this entity lies in the fact that it is a rapidly progressive lymphoma associated with very poor prognosis possibly owing to the association with terminal haemophagocytic syndrome and expression of P-glycoprotein. It is very important to differentiate a composite lymphoma from a T-cell lymphoma with reactive RS-like cells as treatment modalities are very different.





