
Abstract
Polycythaemia vera (PV) is one of the chronic myeloproliferative neoplasms, which are collectively characterised by clonal proliferation of myeloid cells with variable morphologic maturity and haematopoietic efficiency. PV is distinguished clinically from other myeloproliferative neoplasms by the presence of an elevated red blood cell mass because of uncontrolled red blood cell production. This is accompanied by increased white blood cells and platelet production, which is because of abnormal clone of haematopoietic stem cells with increased sensitivity to the different growth factors for maturation. PV can present with variable symptoms because of impaired oxygen delivery caused by slugging of blood, such as headaches, dizziness, vertigo, tinnitus, visual disturbances, and angina pectoris.
Some patients present with bleeding complications (1%); another 1% of patients present with thrombotic complications. It is uncommon for patients with myeloproliferative disorders to present with features of angioedema. There are many reported cases in the literature that describe the relationship between the occurrence of angioedema and lymphoproliferative diseases; however, there are no reported cases describing instances of myeloproliferative neoplasm with angioedema.
In this article, the authors have studied a case of a 53-year-old male who presented with recurrent episodes of features that are suggestive of angioedema. He was diagnosed with JAK2-positive myeloproliferative disorder consistent with PV; this is the first reported case in Jordan.
INTRODUCTION
Polycythaemia vera (PV) is a condition characterised by an increasing number of the red blood cells in the blood.1 It is usually caused by a change in the JAK2 gene; affected bone marrow cells can also develop into other cells found in the blood, which means that patients with PV may also have abnormally high numbers of both platelets and white bloods cells.2 Conditions where the body makes too many of these cells are known as myeloproliferative neoplasms.1 The type of myeloproliferative neoplasm is based on whether too many red blood cells, white blood cells, or platelets are being made. Sometimes the body will make too many of more than one type of blood cell, but usually one type of blood cell is affected more than the others.3
Extra cells in the bloodstream cause the blood to be thicker than normal, which increases the risk for blood clots that can block blood flow in arteries and veins.3 There are several types of myeloproliferative disorders. The most common are PV, essential thrombocythaemia, primary myelofibrosis, and chronic myelogenous leukaemia.4
Annual incidence of PV is estimated at approximately 1/36,000–1/100,000 and prevalence at 1/3,300.5 PV is an acquired disease during a person’s life time; rarely it is inherited in an autosomal dominant pattern.6 PV becomes more common as an individual ages and typically presents for the first time around 60 years old. It is more frequent in males than in females and is associated with mutations in JAK2 and TET2 genes.7
The JAK2 gene provides cell instruction for making JAK2 protein. The JAK2 protein is important for controlling blood cell production in bone marrow from haematopoietic stem cells; it promotes cell division and plays a major role for transmitting signals from outside the cell to the cell’s nucleus through a signalling pathway called JAK–STAT pathway.8
JAK2 mutation is a change of valine to phenylalanine at 617 position (JAK2 V617F). This mutation causes haematopietic cells in the bone marrow to become more sensitive to growth factors such as erythropoietin and thrombopoietin, which leads to over-production of red blood cells, white blood cells, or platelets. This mutation is present in the majority of patients with myeloproliferative neoplasm, nearly 100% of patients with PV, and in approximately 50% of essential thrombocytosis and primary myelofibrosis.9
Because patients with PRV have thicker blood, this can lead to serious health problems such as heart attack or stroke.5 Impaired O2 delivery due to slugging of blood may lead to variable symptoms such as headache, dizziness, vertigo, tinnitus, visual disturbances, angina pectoris, or intermittent claudication.10 In patients with PV, those with a haematocrit target of <45% had a significantly lower rate of major thrombosis than those with a haematocrit target of 45–50%.11
It is uncommon for patients with myeloproliferative disorders, including PV, to present with features of acquired angioedema. Acquired angioedema is due to the acquired deficiency of C1-inhibitor, which was first described in a patient with high-grade lymphoma and is frequently associated with lymphoproliferative diseases.12
C1-inhibitor is a serine protease inhibitor, primarily synthesised by hepatocytes. Its synthesis is upregulated by interferon-γ, IL-6, IL-1, and androgens. C1-inhibitor inhibits the activation of C1R, C1S, activated Hageman factor (x11a), and kallikrein. Kallikreins are proteases that cleave kininogen and release bradykinin. Bradykinin exhibits its effect through activation of bradykinin B2 receptor in the membranes of endothelial and smooth muscle cells, and plays a major role in tissue permeability and vascular dilatation. Elevated blood bradykinin levels are found during clinical flares in patients with angioedema.
There are two types of acquired angioedema: Type I and II. In acquired angioedema Type I, the disorder usually associated with lymphoproliferative malignancies, antibodies or immune complexes are produced that destroy C1-inhibitor function. The most common associated malignancy with this type is B-cell lymphoma. In acquired angioedema Type II, a normal C1-inhibitor molecule is synthesised in adequate amounts but, because of unknown events, autoantibodies to the C1-inhibitor molecule bind to the reactive centre of C1-inhibitor, alter its structure, and impair its regulatory capacity.13 It is unknown which one of these myeloproliferative disorder mechanisms cause acquired angioedema.
There is no consensus as to the optimal therapy for the disorder,14 but treatment is aimed at reducing the chance of the patient developing symptoms and complications. The main treatments are venesection and medication to help slow the production of red blood cells.15 Myelosuppressive drugs can reduce the rate of thrombosis in those patients, but there is concern that their use raises the risk of transformation into acute leukaemia; the drug of choice is hydroxyurea because of its efficacy in preventing thrombosis and low leukaemogenicity.16
Many articles have described the relationship between the occurrence of angioedema and lymphoproliferative diseases, but not myeloproliferative ones. In this article, the authors studied a rare case of a 53-year-old male who presented with recurrent episodes of features suggestive of angioedema, who was diagnosed with PV.
CASE REPORT
A 53-year-old male patient, who was a non-smoker with no known prior medical illnesses, presented to the emergency room with recurrent episodes of swelling of his lips and face. The swelling occurred spontaneously, was not itchy, and disappeared without intervention after a few hours but reoccurred every 5–6 days. His symptoms were associated with mild, diffuse abdominal pain and nausea.
The swelling was not preceded by any drug ingestion or exposure to any irritant, such as plants or dust, and was not accompanied by skin rash. He denied any history of insect bites or previous similar symptoms.
His medication history was reviewed to exclude angiotensin-converting enzyme inhibitor-induced angioedema. He described the feeling of itch after hot baths. He had no shortness of breath, no sore throat, no fever, and no dizziness, and other elements of review of systems were unremarkable.
His physical examination was normal except for the presence of lip and periorbital swelling. His vital signs were stable and his O2 saturation readings were always >94%. Chest, cardiovascular system, abdomen, and lower limb examinations were unremarkable.
A routine complete blood count during his second month of symptoms was undertaken: white blood cells 10.94 million cells/μL; red blood cells 7.97 million cells/μL; haemoglobin 18.8 gm/dL; haematocrit 60%; mean corpuscular volume 75 fL; platelets 709×109/L; neutrophils 70%; lymphocytes 17.2%; monocytes 9.5%; eosinophils 2.7%; basophils 0.5%; erythropoietin level 3 μ/L; and vitamin B12 154 ng/mL.
The erythrocyte sedimentation rate was 2, and antinuclear antibodies were negative. Regarding complement components, C2, C4, thyroid-stimulating hormone, thyroxine, and ferritin were unremarkable. Blood film showed hypochromic, microcytic erythrocytosis with thrombocytosis and neutrophilia.
Findings of bone marrow aspiration were suggestive of myeloproliferative neoplasm with features of PV, complicated by iron deficiency (Figure 1). Microscopic examination of the bone marrow revealed hypercellular bone marrow for age, with an estimated cellularity of 90%. Erythroid precursors were increased, and granulocytic precursors were increased with normal maturation and differentiation. Megakaryocytes were increased, forming clusters with pleomorphic morphology. Reticulin stain showed no marrow fibrosis. Masson’s trichrome stain showed no collagenous fibrosis.
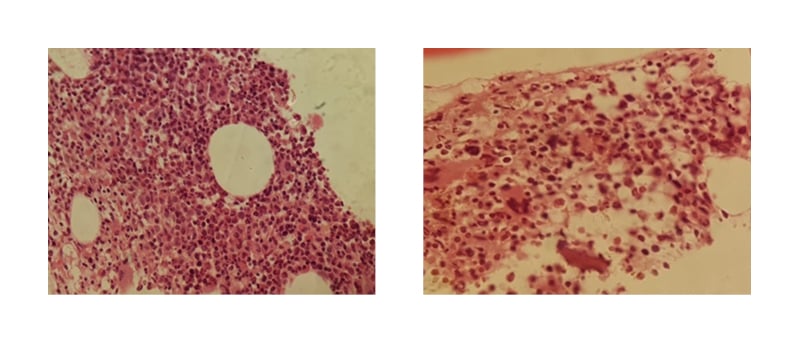
Figure 1: Bone marrow aspiration, showing hypercellular bone marrow with no fibrosis.
Ultrasound of the abdomen revealed a liver measuring 18 cm that was slightly increased in echogenicity. The spleen appeared homogenous, measuring 14 cm.
The patient was positive for JAK2 V617F mutation and negative for BCR-ABL1. His C1 esterase inhibitor level was 0.437 g/L (normal level: 0.15–0.35); however, blood sampling for this test was taken two weeks after commencing hydroxyurea, and not during an acute attack of angioedema, which may have affected the results.
DISCUSSION
The myeloproliferative disorders are clonal disorders of multipotent haematopoietic progenitors and include PV, essential thrombocythaemia, primary myelofibrosis, and chronic myelogenous leukaemia. Most patients with PV, essential thrombocythaemia, and primary myelofibrosis acquire a single point-mutation in the cytoplasmic tyrosine kinase JAK2 (JAK2 V617F).17 There are several criteria to diagnose PV and those criteria depend upon the presence or absence of JAK2 mutation. A previously published study explained the criteria of PV diagnosis:18
- Haemoglobin >16.5 g/dL in males or >16 in females, or haematocrit >49% in males and >48% in females.
- Bone marrow biopsy showing hypercellularity for age.
- Presence of JAK2 V617F mutation.
It was unexpected for a patient with PV to exhibit features of angioedema. Acquired angioedema is characterised by an acquired deficiency of C1-inhibitor, hyperactivation of the classical pathway of human complement, and angioedema symptoms mediated by bradykinin released by inappropriate activation of the contact–kinin system.19 C1-inhibitor acquired angioedema typically presents with recurrent attacks that most commonly involve the tongue, uvula, upper airways, and face, although other areas of the body can be affected.20
There is no cure for PV; treatment focuses on reducing the risk of complications.
Treatment to lower red blood cell levels involves phlebotomy, medications such as hydroxyurea and interferon-α, and radiation treatment, which can help suppress overactive bone marrow cells but raises the risk of leukaemia. Aspirin can relieve bone pain and burning feelings in the hands or feet and reduce the risk of blood clots.21
The discussed patient met all of these criteria for diagnosis of PV. He presented with signs and symptoms of angioedema; although his C1 esterase levels were high, as mentioned the sample was taken after initiation of hydroxyurea and the patient was not experiencing an acute attack, which may have affected the result.
The authors reviewed his medication history to exclude drug-induced angioedema. He denied any history of insect bite or exposure to new food or any allergens. His C4 and C2 levels were normal and there was no family history of angioedema. He responded temporarily to antihistamines and steroids, but once he was commenced on hydroxyurea he did not develop any further episode of angioedema, although his haematocrit remains high, meaning that hydroxyurea may affect the presence of autoantibodies against C1-inhibitor.
The patient was encouraged to exercise regularly, which can improve blood flow and decrease risk of blood clots. He was also advised to avoid low-oxygen environments and extreme temperature.
Underlying lymphoproliferative malignancies are common in patients who are seen initially with late-onset angioedema.22 There are no instances of myeloproliferative neoplasm with angioedema currently reported in the literature and this is the first reported case in Jordan to describe the onset of angioedema as the presenting feature of myeloproliferative disorder; however, further investigations are needed for similar cases. In addition, this highlights the importance of holding high clinical suspicion for neoplasm in adults presenting with angioedema. The authors thank the patient for granting permission to publish this information.





