Abstract
Osteopetrosis (OP) is a rare genetically metabolic bone disorder caused by severe impairment of osteoclast-mediated bone resorption. It is characterised by extensive sclerosis of the skeleton, fragility fracture, haematopoietic insufficiency, nerve entrapment syndromes, and growth impairment. It is clinically classified into two major types: infantile (autosomal recessive, malignant) and adult (autosomal dominant, benign) OP. The infantile type is usually diagnosed early in life, while adult type is diagnosed in late adolescence or adulthood. Approximately one-half of patients are asymptomatic and the diagnosis is made incidentally. However, some patients might present with one or more complications of OP, and the diagnosis is made during the work-up and evaluation. Here, the authors describe an unusual case of adult type OP presented with pancytopenia.
CASE PRESENTATION
The 35-year-old female of Arab ethnicity was admitted to the Department of General Internal Medicine in King Saud Medical City, Riyadh, Saudi Arabia, with a long history of approximately 7 years of intermittent lethargy, dizziness, and exertional dyspnoea. She sought different healthcare centres, where she received multiple blood transfusions, intravenous iron infusion, and vitamins. She gave a history of similar condition to her elder sister, who had passed away. There was no history of bleeding, abnormal menstrual cycle, bony fracture, or haematological disease. On examination, she was fully conscious, alert, and oriented to time, place, and person, with no skeletal deformity. The patient was found to be moderately pale. She had palpable spleen 2 cm below the left subcostal margin in its axis with palpable liver 3 cm below the right subcostal margin, nontender, smooth-surfaced, and soft in consistency. Neurological and ophthalmological examinations were both normal.
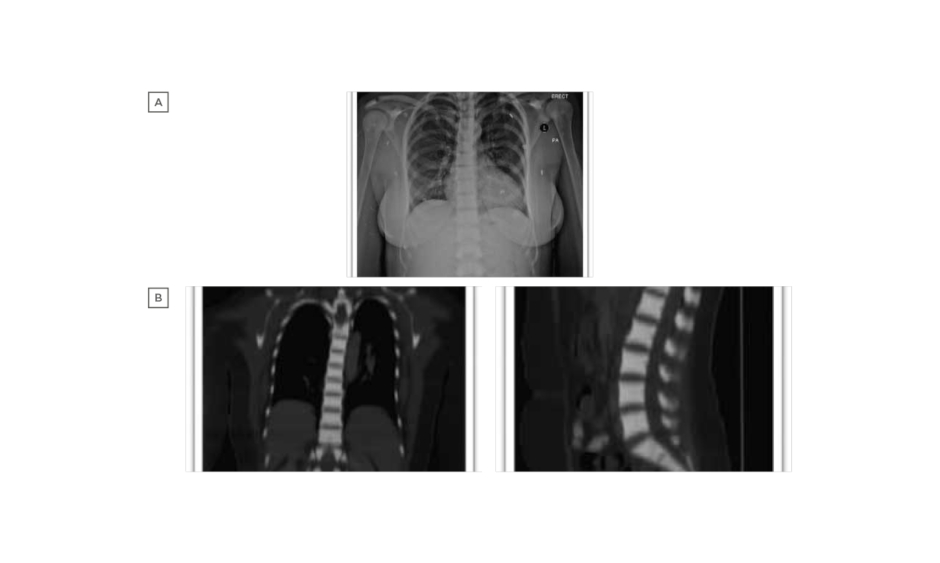
Laboratory examination yielded a red blood cell count 1.44 cells/mm3, haemoglobin 4.80 g/dL, mean corpuscular volume 107.60 fL, mean corpuscular haemoglobin 33.30 pg, red blood cell distribution width 17.70%, reticulocyte count 1.51%, white blood cell counts 2.89 cells/mm3, and platelet count 188.00 /mm3. Peripheral blood smear revealed marked macrocytic anaemia, polychromasia, few teardrop cells, mild leukopenia, and mild thrombocytopenia. Biochemical analysis was normal, including 195 U/L lactate dehydrogenase, total bilirubin level of 9.3 umol/L, and normal serum calcium and phosphorus levels, with the exception of alkaline phosphatase which was 180 U/L. There was a normal iron profile, except for high ferritin which was 631 ng/mL. Thyroid and parathyroid functions were both normal. Serology for cytomegalovirus, Epstein–Barr virus, viral hepatitis, and HIV was negative. The antinuclear antibody titer was nonsignificant, antidouble stranded DNA was negative, and C3 and C4 were both within the normal reference range. Bone marrow aspiration and touch print showed severe hypocellular diluted bone marrow with cellularity <20%. Bone marrow biopsy and microscopic examination showed entirely thick trabecular bone, filling the whole biopsy with marble-shaped bone and no evidence of malignancy. Skeletal survey and radiological evaluation revealed diffuse bone sclerosis (Figure 1). Posteroanterior chest X-ray showed mild cardiomegaly and a homogeneous increase in bone density throughout the clavicular bones, thoracic cage, and vertebrae (Figure 1A). A noncontrast CT scan showed diffuse increased bone density in the vertebral bodies and ribs with severe osseous sclerosis (Figure 1B). Dual-energy X-ray absorptiometry showed generalised increased bone mineral density. The T-score and the age matched Z-score were very high compared to the expected range for the patient’s age according to the International Society of Clinical Densitometry (ISCD) recommendation,1 in keeping with diffuse dense sclerosis noted in previous CT scans (Figure 2).
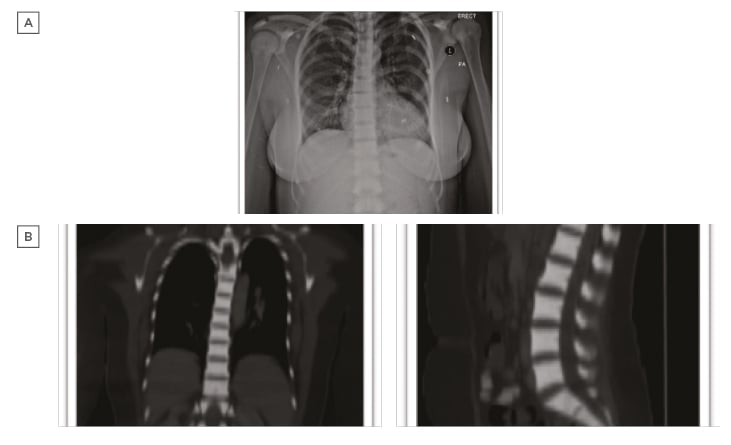
Figure 1: A) Posteroanterior chest X-ray shows mild cardiomegaly and homogeneous increase in bone density throughout the clavicular bones, thoracic cage, and vertebrae. B) Noncontrast CT scan shows diffuse increased bone density in the vertebral bodies and ribs with severe osseous sclerosis
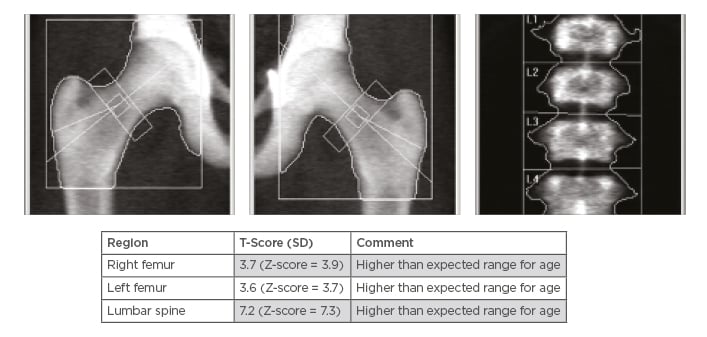
Figure 2: A) Dual-energy X-ray absorptiometry scan using both hips and lumbar spine as standard protocol. B) Dual-energy X-rays Absorptiometry shows generalised increased bone mineral density, and the T-score and the age matched Z-score are very high compared to the expected range for age according to the international Society of Clinical Densitometry (ISCD) recommendations, in keeping with diffuse dense sclerosis noted in previous CT scans.
SD: standard deviation.
Based on the clinical, haematological, radiological, and bone marrow biopsy findings, a final diagnosis of adult-type osteopetrosis (OP) was made. Genetic studies could not be performed. The patient was referred to a specialised centre for bone marrow transplantation where the diagnosis of OP was confirmed. The treating physicians could not find a human leukocyte antigen matching donor in the patient’s family; therefore, the patient was started on corticosteroids as a supportive treatment with regular visits to a haematology outpatient clinic for follow-up clinical and laboratory testing.
DISCUSSION
OP refers to a group of disorders caused by the severe impairment of osteoclast-mediated bone resorption. As a consequence, bone modelling and remodelling are impaired. The defect in bone turnover characteristically results in skeletal fragility despite increased bone mass, and it may also cause haematopoietic insufficiency, nerve entrapment syndromes, and growth impairment. It is clinically classified into infantile, which is autosomal recessive, malignant, and has a poor prognosis; adult, which is autosomal dominant, benign, and has a good prognosis; and intermediate, which is rare, autosomal recessive, and has a poor prognosis.2 OP tarda, the benign adult form, is inherited as an autosomal dominant trait. Patients are typically asymptomatic and have good long-term survival rates because bone marrow failure rarely occurs.3 It is usually detected by a family history of bone disease or as an incidental radiologic finding, and is asymptomatic in approximately 50% of cases. Close to 40% of patients present with fractures related to brittle osteopetrotic bones or with osteomyelitis, especially of the mandible. There is sufficient retention of marrow cavity for normal haematopoiesis to occur in patients with OP tarda. In some cases, there is an elevated acid phosphatase level. Although patients with OP tarda have an increased susceptibility to fractures, healing appears to proceed normally.4
OP is characterised by abnormal bone tissue which infiltrates the marrow cavity, resulting in pancytopenia and bone marrow failure. As a compensatory mechanism, extramedullary haematopoiesis occurs in the spleen and/or liver. Clinical features include recurrent fractures, stunted growth, failure to thrive, blindness and deafness from cranial nerve entrapment, and recurrent infections from impaired immune function. These severe clinical features of OP are more common in infantile than adult OP.5
In this case, severe pancytopenia was the patient’s main presentation with a long history of the symptoms of anaemia and multiple blood transfusions, which is an unusual presentation of adult OP. Moreover, there was no previous history of bony fracture. One of the patient’s elder sisters had similar symptoms, but she died with no clear diagnosis and no family history of OP. She also had hepatosplenomegaly upon palpation, which is likely because of bone expansion leading to bone marrow narrowing and extramedullary haematopoiesis.
The diagnosis of OP largely depends on the radiologic features of the skeleton. The presence of bone sclerosis is usually diagnostic of OP. Two types of adult OP are mostly identified based on the distribution of bone sclerosis. Typing the patient’s disease helps to predict a fracture pattern because of the nature of the different types. In Type I, sclerosis of the skull mainly affects the vault and the spine does not show much sclerosis. Type II tends to increase the risk of fracture, and sclerosis is mainly found in the base of the skull, the spine always has the rugger jersey appearance, and the pelvis always shows subcristal sclerosis. Transverse banding of metaphysis is common in patients with Type II disease but not in patients with Type I. This finding confirms Type II disease, but its absence does not necessarily indicate Type I.2
Some controversy surrounds the role of bone marrow biopsy in the diagnosis of OP; however, the histopathologic studies of the biopsy may be useful to predict the likelihood of bone marrow transplant success. Patients with crowded bone marrow are less likely than others to respond to a transplant. For this patient, the diagnosis of OP was established based on the radiological evidence of dense sclerosis, and bone marrow biopsy was performed to confirm its diagnosis and rule out other causes of pancytopenia.
Genetic studies could not be performed for the patient. A defect in chloride channel 7 α subunit (CLCN7) has been identified in some cases of infantile and adult OP. Most individuals diagnosed with autosomal dominant CLCN7-related OP have an affected parent. The proportion of cases caused by de novo pathogenic variants is unknown. Each child of an individual with autosomal dominant CLCN7-related OP has a 50% chance of inheriting the pathogenic variant.6
The treatment of OP depends on its variety and presence of complications. Most cases of adult OP require no specific treatment by itself. This patient, however, had severe pancytopenia, and therefore was referred to a specialised centre for haematopoietic stem cell transplantation, of which might be the only treatment that can provide a cure in such patients. IFNγ1b treatment has been tried in patients with OP and has been reported to result in improved immune function, increased bone resorption, and increased bone marrow space.7-9 At the time of the study, there was insufficient evidence to support the routine use of corticosteroids in OP.10
CONCLUSION/TAKE-HOME MESSAGES
The take-home message of this case is to maintain a high index of clinical suspicion for unusual presentation of adult-onset OP. Therefore, proper clinical assessment and radiographic investigations are essential for diagnosis of OP. Thus, it is important for physicians to be familiar with various clinical manifestations of OP and its radiological features.





